Google DeepMind dévoile AlphaGenome, un puissant modèle d’apprentissage profond pouvant prédire les impacts de variants ou de mutations génétiques sur un large éventail de processus biologiques. L’outil d’IA permettrait de prédire avec précision les fonctions de longs brins d’ADN allant jusqu’à un million de paires de bases, permettant ainsi de mieux comprendre les fonctions des gènes à l’échelle moléculaire ainsi que leur implication dans les maladies génétiques.
Les variants génétiques influencent les processus biologiques et peuvent modifier la réponse d’un organisme à son environnement ou sa sensibilité aux maladies. Ils peuvent aussi contribuer à l’apparition de maladies génétiques. Interpréter ou prédire la manière dont ces modifications affectent les fonctions du génome demeure cependant l’un des plus grands défis en sciences biomédicales.
En effet, les impacts des variants non codants (des séquences qui ne codent pas pour des protéines mais qui peuvent influencer l’expression des gènes) sont particulièrement difficiles à prédire en raison de la diversité des effets moléculaires qu’ils peuvent induire. Par exemple, les variants non codants peuvent moduler des propriétés du génome telles que l’accessibilité de la chromatine (le complexe d’ADN, d’ARN et de protéines constituant les chromosomes), les modifications épigénétiques et la conformation tridimensionnelle de la chromatine.
Dans ce contexte, les variants peuvent également influencer la disponibilité d’ARN messager (ARNm) et peuvent avoir des effets spécifiques selon le type cellulaire ou tissulaire. Or, plus de 98 % des variants chez les humains se produisent dans les régions non codantes, ce qui rend difficile la prédiction de leurs impacts. Des modèles informatiques sont utilisés pour la prédiction de ces effets, mais ceux existants ne peuvent prédire avec précision les impacts des variants à longues séquences.
Les chercheurs de Google DeepMind ont développé AlphaGenome pour effectuer des prédictions à haute résolution sur de longues séquences d’ADN. « Nous présentons AlphaGenome, un nouvel outil d’intelligence artificielle (IA) qui prédit de manière plus complète et précise l’impact de variants ou de mutations uniques dans les séquences d’ADN humain sur un large éventail de processus biologiques régulant les gènes. Ceci a été rendu possible, entre autres, par des avancées techniques permettant au modèle de traiter de longues séquences d’ADN et de produire des prédictions de haute résolution », expliquent dans un communiqué Ziga Avsec et Natasha Latysheva, les codéveloppeurs de l’outil.
« Nous pensons qu’AlphaGenome peut être une ressource précieuse pour la communauté scientifique, aidant les scientifiques à mieux comprendre la fonction du génome, la biologie des maladies et, en fin de compte, à stimuler de nouvelles découvertes biologiques et le développement de nouveaux traitements », ont-ils ajouté.
Un modèle entraîné sur des millions de signaux biologiques
D’après les chercheurs, AlphaGenome peut prendre en charge de longues séquences d’ADN allant jusqu’à un million de paires de bases et peut prédire des milliers de propriétés moléculaires. Il a été entraîné à partir d’un vaste ensemble de données publiques incluant des expériences évaluant les effets de ces propriétés dans des cellules et tissus humains et murins. Plusieurs unités de traitement tensoriel (des circuits intégrés spécifiques développés par Google pour optimiser l’apprentissage automatique) interconnectées ont été utilisées pendant l’entraînement.
Parmi les propriétés qu’il peut prédire figurent le début et la fin des séquences génétiques dans différents types cellulaires et tissulaires, les sites d’épissage (des régions où une partie de l’ADN ou de l’ARN est sectionnée, puis les deux extrémités restantes sont réassemblées), la quantité d’ARN produite, ainsi que les bases d’ADN accessibles, proches les unes des autres ou liées à certaines protéines.
Le modèle serait capable de prédire simultanément 5 930 signaux génétiques humains et 1 128 signaux génétiques murins liés à des fonctions spécifiques. Il permettrait d’évaluer les effets des variants génétiques ou des mutations en comparant les prédictions des séquences mutées à celles des séquences non mutées.
« En permettant une prédiction à haute résolution pour les longues séquences d’entrée, AlphaGenome peut prédire la gamme de modalités la plus diversifiée. Ce faisant, AlphaGenome fournit aux scientifiques des informations plus complètes sur les étapes complexes de la régulation des gènes », expliquent Avsec et Latysheva.
Des maladies génétiques rares, telles que l’amyotrophie spinale et certaines formes de mucoviscidose, peuvent être causées par des erreurs d’épissage de l’ARN. AlphaGenome aurait permis de modéliser avec précision la localisation et le niveau d’expression de ces modifications.

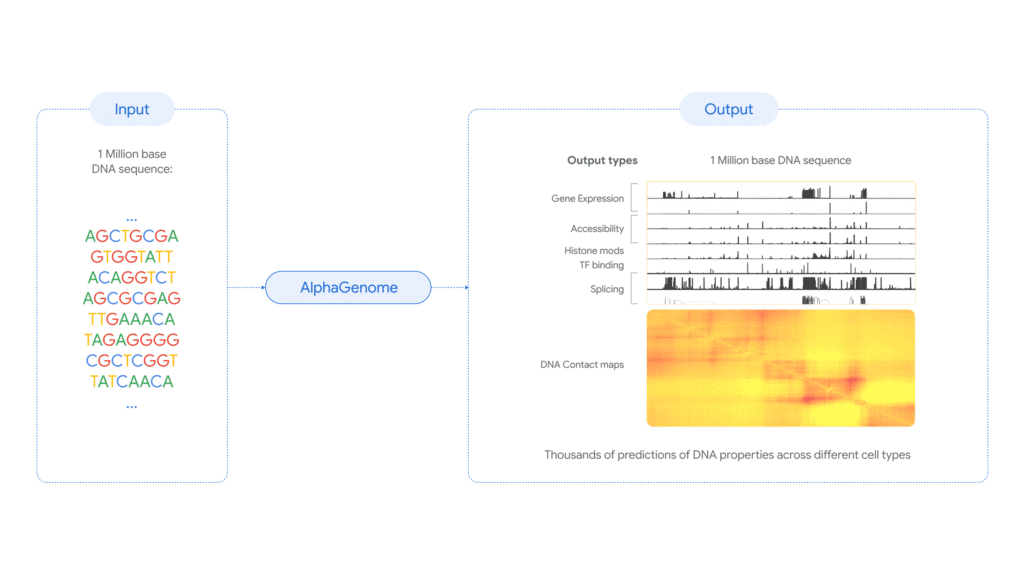 Graphique résumant le mode de fonctionnement d’AlphaGenome. © Google DeepMind
Graphique résumant le mode de fonctionnement d’AlphaGenome. © Google DeepMindDes performances qui rivalisent avec les meilleurs modèles existants
Les tests de performance – détaillés dans l’étude publiée aujourd’hui dans la revue Nature – ont montré qu’AlphaGenome a rivalisé, voire surpassé, les meilleurs modèles existants dans un large éventail de prédictions génomiques.
Par exemple pour la prédiction de séquences d’ADN uniques, il a surpassé les modèles de pointe existants dans 22 évaluations sur 24. Pour la prédiction de l’effet régulateur d’une variante génétique, il a égalé ou surpassé les modèles externes les plus performants dans 24 évaluations sur 26.
L’équipe a également testé le modèle pour explorer les mécanismes déclenchés par une mutation associée à la leucémie lymphoblastique aigue à cellules T. Les études antérieures sur la maladie ont mis au jour des mutations au niveau de régions spécifiques du génome. AlphaGenome a prédit que ces mutations pourraient activer un gène voisin appelé TAL1 en introduisant un motif de liaison spécifique à l’ADN.


« AlphaGenome sera un outil puissant dans ce domaine », explique Marc Mansour de l’University College of London, dans le communiqué. « Déterminer la pertinence des différentes variantes non codantes peut s’avérer extrêmement complexe, notamment à grande échelle. Cet outil apportera une pièce essentielle du puzzle, nous permettant d’établir de meilleurs liens pour comprendre des maladies comme le cancer. »
Malgré ces performances, le modèle présente toutefois encore certaines limites. Bien qu’il puisse par exemple prédire les effets moléculaires des variantes génétiques, il n’offre pas d’aperçu complet de la manière dont elles conduisent à l’apparition de traits ou de maladies complexes. Ces derniers impliquent notamment des processus biologiques plus larges tels que des facteurs développementaux et environnementaux.
Des améliorations pourraient néanmoins être apportées, notamment en augmentant significativement les données d’entraînement, le nombre d’espèces couvertes, ainsi que la gamme des séquences non codantes reconnues par le modèle. AlphaGenome est disponible pour une utilisation non commerciale pour que les chercheurs du monde entier puissent le tester de leur côté et faire part de leurs commentaires quant aux améliorations à apporter.
Source : Nature
Vidéo de présentation de l’outil :
