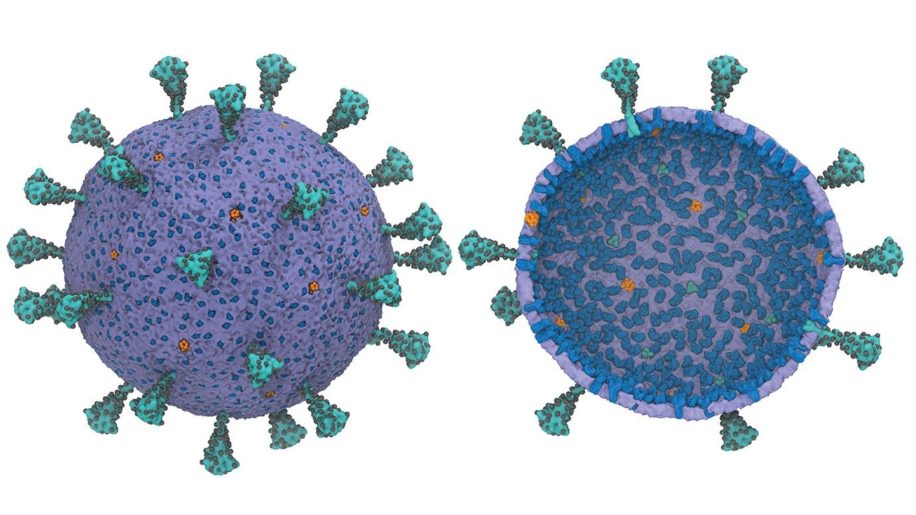
Dans toute lutte contre un virus, les premières étapes sont de visualiser au mieux et comprendre les mécanismes d’infection. Depuis les débuts de la pandémie actuelle, les chercheurs se sont montrés extrêmement rapides et efficaces pour séquencer le génome du coronavirus, modéliser sa protéine Spike et étudier les différentes étapes de l’infection, ce qui a permis notamment de commencer à développer des vaccins à une vitesse record.
Cependant, bien que des modèles de la protéine Spike (permettant au virus de se fixer aux cellules hôtes) aient déjà été publiés l’année dernière, jusqu’ici aucun modèle informatique complet et exploitable du SARS-CoV-2 entier n’avait vu le jour. Récemment, des chercheurs de l’Université de Chicago ont créé le premier modèle informatique (CG) utilisable de l’ensemble du virus, qu’ils mettent d’ores et déjà à la disposition de tous pour faire avancer la recherche. Le modèle et les méthodes qui en résultent permettent maintenant la simulation des virions du SARS-CoV-2.
« Si vous pouvez comprendre comment un virus fonctionne, c’est la première étape pour l’arrêter », a déclaré le professeur Gregory Voth, chercheur en informatique et responsable de l’équipe qui a développé le modèle, détaillé dans la revue Biophysical Journal. « Chaque chose que nous obtenons sur le cycle de vie et la composition du virus est un point de vulnérabilité que nous pouvons cibler et atteindre ».
Pour développer leur modèle, Voth et son équipe se sont appuyés sur leur expérience antérieure pour identifier les caractéristiques les plus importantes de chaque composant individuel du virus et ont laissé de côté les informations « moins importantes », afin d’aboutir à un modèle informatique simple mais complet, et surtout exploitable pour la recherche. Cette technique est appelée « coarse-graining », que Voth et ses étudiants ont contribué à mettre au point.
La structure simplifiée obtenue permet d’aborder une question clé de la recherche en matière de santé : même si un virus est l’une des entités biologiques les plus simples, la modélisation informatique reste un défi majeur, surtout si l’on veut simuler l’une des interactions d’un virus avec le corps de son hôte, ce qui signifierait représenter des milliards d’atomes…
Un défi informatique et biologique de taille
« Vous pourriez essayer d’exécuter un modèle au niveau de l’atome du virus entier, mais sur le plan informatique, cela vous bloquerait immédiatement », a déclaré Voth. « Un ordinateur pourrait le gérer assez longtemps pour modéliser disons quelques centaines de nanosecondes de mouvement, mais ce n’est vraiment pas assez long pour obtenir les informations les plus utiles ».
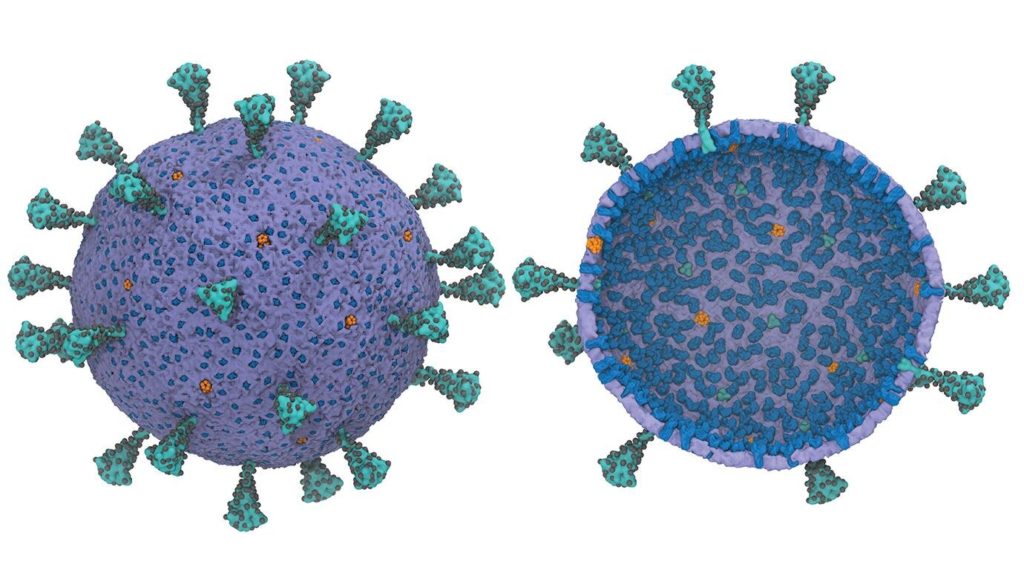
Ainsi, de nombreux chercheurs se sont concentrés sur la création de modèles de protéines individuelles du virus. Mais Voth explique que bien ce processus segmenté a ses utilités, il manque également une partie du tableau d’ensemble… « Le virus lui-même est un système holistique », déclare Voth. « À mon avis, on ne peut pas supposer que l’on peut comprendre les différentes parties de manière isolée. Les virus sont bien plus que la simple somme de leurs parties », ajoute le professeur de chimie Haig P. Papazian.
Le laboratoire de Voth travaille depuis des années à la modélisation d’autres virus, comme le VIH. L’une des leçons qu’ils ont apprises est que de multiples parties du virus travaillent en coopération, et non de façon isolée. Par exemple, en visualisant les mécanismes du virus dans son ensemble, les scientifiques peuvent mieux étudier la conception d’un médicament qui se lie aux protéines de pointe à la surface du virus pour les empêcher de s’attacher aux cellules hôtes. « L’une des principales questions que vous pourriez vous poser alors est de savoir s’il faut doser chaque protéine de pointe pour qu’elle fonctionne. Si ce n’est pas le cas, quel est le pourcentage le plus faible que vous pouvez obtenir ? », déclare Voth. « C’est une question clé lorsque vous essayez de créer des médicaments ou des anticorps, et c’est quelque chose que vous pouvez mieux comprendre en étudiant le virus dans son ensemble ».

Dans cette vidéo, on y voit le SARS-CoV-2 modélisé bouger selon l’un des algorithmes de mouvement établis par l’équipe :
Ce nouveau modèle informatique du SARS-CoV-2 fournit également un cadre dans lequel les chercheurs pourront intégrer des informations supplémentaires dès que de nouvelles découvertes seront faites. Voth et ses collègues espèrent qu’il s’avérera utile pour la conception de médicaments contre d’autres coronavirus également ainsi que pour comprendre les mutations qui peuvent survenir, comme celles récemment détectées au Royaume-Uni et en Afrique du Sud.
« Créer un modèle multi-échelle de l’ensemble du virus et intégrer rapidement toutes ces informations est un grand pas en avant technologique », déclare Voth. « Je suis vraiment fier de mon laboratoire. Nous l’avons fait en un temps record, vraiment – quelques mois seulement. S’il y a un aspect positif à cette pandémie, j’espère qu’elle fera progresser nos outils de lutte contre les virus au-delà de celui responsable de la COVID-19, du VIH et contre tout nouveau coronavirus qui pourrait apparaître à l’avenir ».
Par le passé, les simulations informatiques (CG) des processus viraux ont permis d’élucider un large éventail de mécanismes pour différentes maladies. Par exemple, dans le cas du VIH, les simulations CG ont contribué à la compréhension de l’auto-assemblage de la capside et de la reconnaissance et du blocage de l’activité virale par les récepteurs du système immunitaire inné, ainsi que de son inhibition par diverses molécules médicamenteuses. « Il est probable que des sondes moléculaires dans les processus impliquant un modèle holistique du virion du SARS-CoV-2 puissent aider à révéler de nouvelles voies pour combattre le virus en exploitant des mécanismes viraux impliquant un comportement à grande échelle », écrivent les chercheurs dans le document.