
Il y a 20 ans, des chercheurs pensaient enfin y être arrivés, séquençant ce qu’ils définissaient comme l’ensemble du génome humain. Cependant, ils se sont aperçus que des bribes de code génétique étaient encore manquantes. Aujourd’hui, après des décennies de recherches et développement, ils sont enfin parvenus à combler ces lacunes et obtenir le tout premier séquençage complet du génome de notre espèce.
Cet exploit a notamment été possible grâce aux récentes avancées au niveau des technologies et méthodes de lecture de l’ADN, qui ont fait une grande différence. « Ayant participé au premier projet sur le génome humain en 2001, et m’étant particulièrement concentré sur les régions difficiles, je suis très satisfait de voir ce projet réalisé, même si cela a pris 20 ans », déclare Evan Eichler, de l’université de Washington à Seattle.
Le nouveau génome comprend 200 millions de paires de bases, ou « lettres » d’ADN supplémentaires, et ajoute plus de 2000 gènes au précédent modèle. Nos gènes contribuent à faire de nous ce que nous sommes. Les humains en possèdent des milliers, bien que le nombre exact soit incertain et dépende en partie de la façon dont on les compte. Ils sont stockés sur de longues molécules d’ADN au centre des cellules. L’information génétique existe sous la forme de quatre molécules appelées bases (C, G, T et A) qui sont présentes le long de la molécule d’ADN.
Le génome humain contient ainsi un peu plus de 3 milliards de « lettres ». Les premières séquences « complètes » ont été publiées en 2001 : l’une par le Consortium international de séquençage du génome humain (HGSC) et l’autre par la société américaine Celera Genomics. Le projet avait débuté dix ans plus tôt, en 1990. Comme le génome devait être lu par petits morceaux puis réassemblé, certaines sections hautement répétitives se sont avérées impossibles à placer, un peu comme un puzzle dont toutes les pièces se ressemblent.
Combler les lacunes
Au cours des trois années suivantes, le HGSC a comblé certaines des lacunes et, en 2004, le consortium a annoncé qu’il avait fait tout ce qu’il pouvait. Les généticiens ont continué à améliorer le génome de référence, mais en grande partie en améliorant la précision des séquences existantes, plutôt qu’en en ajoutant de nouvelles. Environ 8% des séquences étaient encore soit manquantes, soit susceptibles d’être erronées.
La nouvelle version du génome a été créée par le consortium Telomere-to-Telomere, dirigé par Karen Miga, de l’université de Californie à Santa Cruz, et Adam Phillippy, du National Human Genome Research Institute dans le Maryland. En 2018, ils ont fait partie d’une équipe qui a séquencé de gros morceaux du génome, de plus de 100 000 bases. Cela leur a permis de combler certaines parties manquantes. « Elle [Miga] m’a appelé et m’a dit ‘Je veux terminer le génome’ », raconte Phillippy. J’ai répondu : « Moi aussi ».
Les deux chercheurs ont alors choisi de lire l’ADN d’une lignée cellulaire appelée CHM13. Elle provient d’une masse de tissu appelée môle hydatiforme, une sorte de grossesse ratée dans laquelle un ovule dans un utérus a en quelque sorte perdu son génome, et a ensuite été fécondé par un spermatozoïde. La cellule résultante n’avait que la moitié de l’ADN d’un embryon normal, et l’ADN du spermatozoïde a donc été dupliqué. De telles cellules forment des excroissances dangereuses, pouvant donner naissance à des cancers, et doivent être retirées. Elles peuvent ensuite être cultivées en laboratoire.
« C’est un cas unique, car il ne s’agit pas du génome d’une personne ayant vécu », explique Phillippy. L’ADN provient d’un seul spermatozoïde, il s’agit donc de la moitié du génome d’un père potentiel, qui a été dupliqué. Les cellules ont été recueillies de manière consensuelle il y a plusieurs décennies, mais l’identité du donneur a été rendue anonyme par une société qui a depuis cessé ses activités, de sorte que l’on ne sait pas de qui elles proviennent. « Nous ne pouvons pas vraiment savoir, même si nous le voulions, de qui elles proviennent à l’origine », déclare Phillippy.
Les cellules humaines normales possèdent deux copies de chaque portion d’ADN, qui présentent souvent des différences importantes car l’une provient de la mère et l’autre du père. Il est donc plus difficile de séquencer l’ADN avec précision, car il est difficile de distinguer les erreurs de séquençage des véritables différences. L’utilisation de CHM13 permet d’éviter ce problème, car les deux copies sont pratiquement identiques.

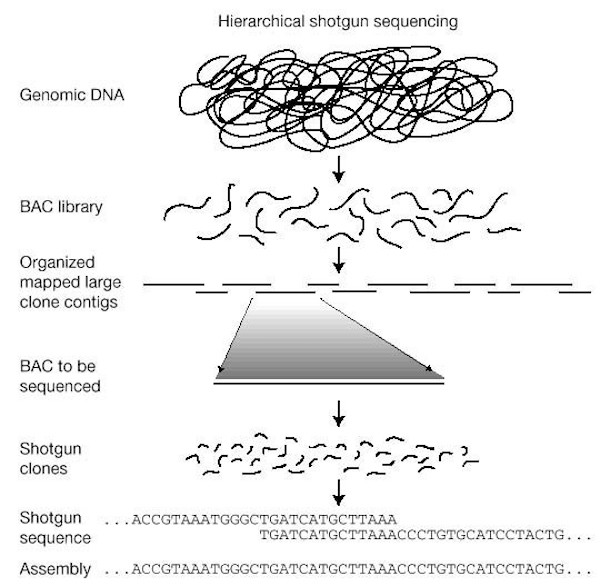 Une « bibliothèque génétique » est construite en fragmentant le génome cible et en le clonant dans un vecteur de clonage à grands fragments ; ici, les vecteurs BAC (chromosomes artificiels bactériens) sont représentés. Les fragments d’ADN génomique représentés dans la banque sont ensuite organisés en une carte physique et les clones BAC individuels sont sélectionnés et séquencés par la stratégie du « random shotgun ». Enfin, les séquences des clones sont assemblées pour reconstruire la séquence du génome. © International Human Genome Sequencing Consortium
Une « bibliothèque génétique » est construite en fragmentant le génome cible et en le clonant dans un vecteur de clonage à grands fragments ; ici, les vecteurs BAC (chromosomes artificiels bactériens) sont représentés. Les fragments d’ADN génomique représentés dans la banque sont ensuite organisés en une carte physique et les clones BAC individuels sont sélectionnés et séquencés par la stratégie du « random shotgun ». Enfin, les séquences des clones sont assemblées pour reconstruire la séquence du génome. © International Human Genome Sequencing ConsortiumDes techniques de séquençage complémentaires
Pour assembler les séquences du génome, l’équipe a combiné deux technologies. L’une était un type de séquençage qui lit des tronçons extrêmement longs, de plus d’un million de lettres, et l’autre un type qui offre une précision extrêmement élevée et peut donc traiter des sections très légèrement différentes — comme les copies multiples d’un même gène.
L’ADN humain est stocké sur de grandes molécules appelées chromosomes, dont les quatre bras se rejoignent en leur centre pour former un X. Une grande partie de l’ADN difficile à lire se trouve autour des points centraux, appelés centromères. En outre, certains chromosomes sont asymétriques, avec une paire de bras plus courts que l’autre : les bras courts contiennent beaucoup d’ADN « difficile à lire ».
En août 2020, l’équipe a publié le chromosome XY humain complet, déterminant le sexe. Ils ont maintenant publié l’intégralité du génome humain. La nouvelle version ajoute près de 200 millions de lettres à la version précédente, avec 2226 sections qui sont des copies quasi identiques de gènes connus. Parmi ces nouveaux gènes, l’équipe prévoit que 115 codent pour des protéines. La série de trois articles détaillant cette avancée a été publiée sur le serveur de préimpression bioRxiv.
Comprendre ce qu’est un gène…
Cependant, Phillippy souligne que ces chiffres sont incertains… « La définition de ce qu’est un gène est encore un peu confuse », dit-il. Les gènes sont traditionnellement considérés comme des sections d’ADN qui codent pour une protéine, mais en réalité, de nombreux gènes sont non codants et ont d’autres fonctions. Le nouveau génome compte 63 494 gènes, contre 60 090 dans la dernière mise à jour effectuée en 2019. Les gènes qui codent les protéines sont au nombre de 19 969, contre 19 890. « C’est beaucoup, beaucoup mieux que tout ce que nous avions », déclare Aida Andrés de l’University College London.
Dans un deuxième article, l’équipe d’Eichler s’est concentrée sur les duplications segmentaires : de longues portions d’ADN qui ont été copiées encore et encore. Contrairement à « l’ADN poubelle », qui est souvent constitué de répétitions apparemment dénuées de sens, les duplications segmentaires comprennent des gènes et d’autres séquences qui ont des fonctions reconnaissables. Grâce à elles, les individus peuvent avoir de nombreuses copies de certains gènes.

Les duplications segmentaires représentent près d’un tiers de la nouvelle séquence et constituent 7 % du génome. Leurs séquences varient également plus que les régions non dupliquées. Eichler pense que les duplications segmentaires ont joué un rôle clé dans l’évolution humaine. « Elles sont l’endroit du génome où de nouveaux gènes sont susceptibles de naître », explique-t-il, car l’une des copies est libre de varier. Les humains possèdent plusieurs gènes dupliqués, qui, selon lui, semblent avoir été « essentiels au développement d’un plus grand cerveau, qui nous distingue des autres primates ».
Selon Andrés, même si les gènes dupliqués ne deviennent pas significativement différents, ils peuvent néanmoins avoir des effets profonds s’ils signifient simplement qu’une protéine est fabriquée en plus grande quantité. Selon elle, les duplications segmentaires ne peuvent pas expliquer toute l’évolution humaine, car il s’agit certainement d’un processus extrêmement complexe, « mais elles sont importantes ».
Activer et désactiver l’ADN
Toujours selon Andrés, le nouveau génome facilitera grandement l’étude des gènes dupliqués, car les séquences qu’il répertorie sont beaucoup plus susceptibles d’être correctes que les versions antérieures. Il est crucial de comprendre les duplications segmentaires car certaines d’entre elles sont à l’origine de troubles génétiques, explique Eichler.
Dans un troisième article, une équipe dirigée par Winston Timp de l’université Johns Hopkins de Baltimore a examiné des marqueurs chimiques appelés groupes méthyles qui s’attachent à l’ADN en divers points. Ces marqueurs « épigénétiques » affectent les gènes qui sont activés ou désactivés. L’équipe de Timp a utilisé le nouveau génome pour cartographier la méthylation dans les zones nouvellement explorées.
Les chercheurs ont alors constaté que les niveaux de méthylation sont faibles autour des centromères, au cœur des chromosomes. Ces régions sont cruciales pour la reproduction et la division cellulaire. Lorsque cela se passe mal, les résultats peuvent être dangereux. « Dans le cas du cancer, il arrive souvent que l’on gagne un chromosome entier ou que l’on en perde un », explique Timp. À long terme, la compréhension du fonctionnement de la division cellulaire et du rôle que la méthylation pourrait jouer pourrait ouvrir la voie à de nouveaux traitements contre le cancer.
