Identifier les causes et mécanismes précis sous-tendant le développement des maladies est primordial afin de développer des solutions thérapeutiques plus efficaces et précises. Jusqu’à présent, les scientifiques ne se concentraient que sur l’identification individuelle de facteurs en examinant les processus pathologiques un par un. Récemment, une équipe de chercheurs a développé une technique utilisant l’intelligence artificielle afin de comparer simultanément l’activité génomique de plusieurs maladies afin d’identifier des facteurs jusqu’alors inconnus.
Les chercheurs de l’Université de Princeton ont franchi une étape cruciale concernant les causes et les caractéristiques des maladies en mettant à profit l’apprentissage automatique pour analyser les modèles moléculaires de centaines de maladies simultanément.
Démontrant l’utilisation d’un nouvel outil maintenant disponible pour les chercheurs du monde entier, l’équipe d’informaticiens et de biologistes a déjà découvert et confirmé expérimentalement des contributions auparavant inconnues de quatre gènes à une forme rare de cancer qui affecte principalement les bébés et les jeunes enfants.
L’équipe, qui comprend des collaborateurs de la Michigan State University et de l’Université d’Oslo, a présenté le système et démontré ses capacités dans un article publié dans la revue Cell Systems. Alors que les approches précédentes mettaient l’accent sur les gènes associés à des maladies ou à des types de cancer spécifiques, la nouvelle technique utilise l’apprentissage automatique pour trouver des modèles uniques d’activité des gènes en examinant simultanément plus de 300 maladies différentes.
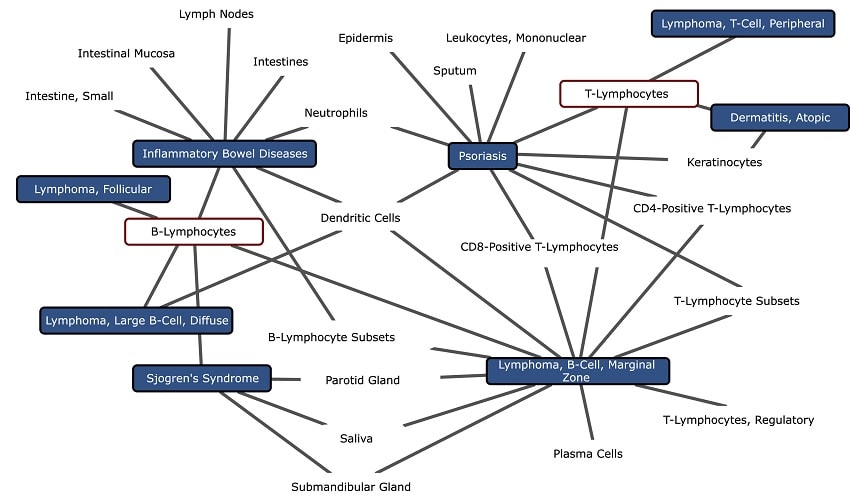
Notamment les cancers, les maladies cardiaques, les troubles métaboliques et bien d’autres. Ce faisant, elle révèle des distinctions entre les maladies et les types de tissus, y compris des différences précises entre des maladies apparentées qu’il était impossible de discerner avec d’autres techniques. Les chercheurs pensent que, avec le développement ultérieur, l’outil sera utile aux cliniciens pour diagnostiquer les maladies, adapter et suivre l’efficacité des traitements ainsi que trouver de nouvelles approches thérapeutiques.
Comparer simultanément plusieurs gènes pour plus d’efficacité
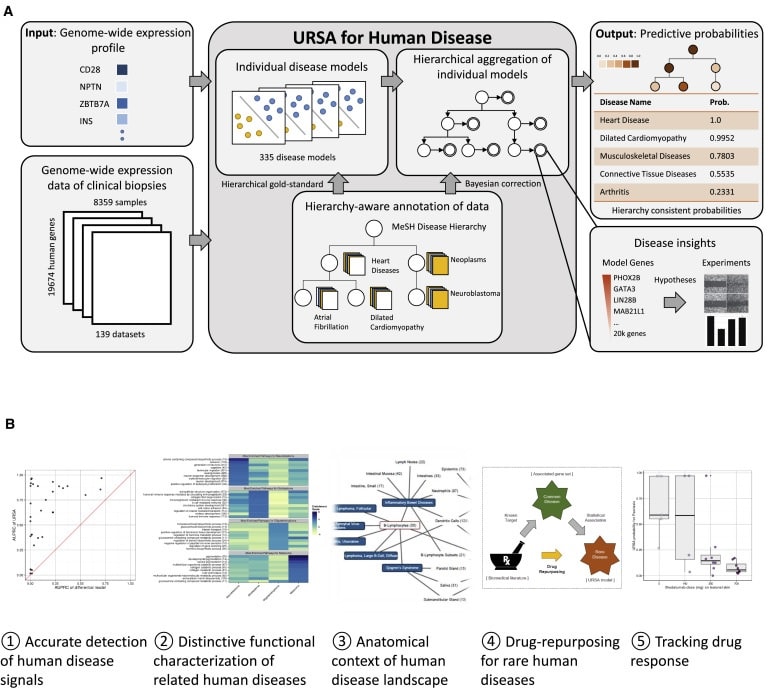
Le système, appelé Unveiling RNA Sample Annotation for Human Diseases, ou URSA (HD), incorpore des informations sur l’activité des gènes provenant d’enregistrements disponibles au public d’environ 8000 biopsies prélevées sur des tissus sains et malades de milliers de patients. À l’avenir, les chercheurs pourront soumettre de nouveaux échantillons à l’outil via une interface Web et recevoir une analyse des associations possibles avec des maladies et des types de tissus.
« La véritable innovation consiste à comparer tous les échantillons à un échantillon sur deux » déclare Chandra Theesfeld. Theesfeld a comparé l’idée à la capacité des humains à reconnaître des différences nuancées entre les comportements en se basant sur une grande variété d’exemples. Regarder des joueurs de football, par exemple, peut révéler les caractéristiques d’une action du pied, mais regarder des joueurs de football et des danseurs de ballet en même temps révèle les détails et le contexte d’une action similaire avec un style et un but très différents.
« Les étudier ensemble permet de distinguer des aspects uniques ». Ce point de vue offre un moyen non biaisé « d’apprendre de nouvelles choses sur la maladie qu’il est impossible de trouver avec l’approche par maladie individuelle — et potentiellement d’identifier de nouvelles cibles pour des traitements ou même de découvrir de nouveaux aspects de la maladie, qui étaient alors inconnus ».
En faisant ces comparaisons, l’algorithme accorde plus de poids aux différences d’activité des gènes qui définissent de manière unique les tissus et les maladies distincts. Il met moins l’accent sur l’information de l’activité des gènes commune aux maladies apparentées, dont une grande partie est déjà bien étudiée.
Sur le même sujet : Babylon : l’intelligence artificielle qui diagnostique des patients aussi bien que des médecins expérimentés
Dans l’analogie football-danse, c’est comme mettre de côté l’action à grande échelle consistant à lever une jambe dans un coup de pied et à trouver de nombreux détails, tels que l’angle du pied, qui, observés ensemble, constituent un ensemble particulier de caractéristiques qui identifient de manière fiable une action.
« Notre méthode repose sur les informations relatives à la maladie de l’échantillon de patients. Elle n’est donc pas biaisée en faveur des gènes populaires de la maladie, qui font toujours l’objet d’études » explique Theesfeld. « Nous pouvons suivre l’évolution des données sans savoir exactement ce que chaque changement signifie ».
Créer des signatures génomiques caractéristiques en examinant l’ARN
Theesfeld a noté que 90% des études sur les gènes ne portent que sur 10% des gènes humains. URSA (HD) examine le génome humain dans son ensemble et crée un modèle ou une signature génomique pour chaque maladie. Cette approche pourrait être particulièrement efficace pour les maladies rares, pour lesquelles les chercheurs peuvent désormais créer un modèle avec seulement quelques échantillons.

Dans le cas du neuroblastome, un cancer pédiatrique, les chercheurs ont découvert quatre gènes qui contribuaient particulièrement à la maladie, et pour lesquels il n’existait aucune information préalable dans la littérature scientifique. Pour confirmer les résultats, Theesfeld a effectué des tests de laboratoire sur des cellules humaines, manipulant l’activité des gènes et observant leurs effets sur les processus liés au cancer dans les cellules.
Plutôt que de regarder l’ADN lui-même, URSA (HD) examine l’ARN, le produit que les cellules créent en transcrivant l’information contenue dans l’ADN en molécules actives qui construisent et dirigent des cellules, et transmettent des signaux d’une cellule à l’autre.
De cette manière, le système va au-delà des mutations (brouillage dans les gènes eux-mêmes) et se concentre plutôt sur les produits de transcription en aval, qui peuvent être dérégulés de manière à causer des problèmes même si le gène d’origine est normal.
La recherche fait partie d’un travail de longue date dans le laboratoire de Troyanskaya visant à intégrer d’énormes collections d’ensembles de données dissemblables afin d’extraire les informations nécessaires pour effectuer des prévisions biologiques précises et pour diriger des expériences de laboratoire afin d’accélérer la découverte.
Un large éventail de données scientifiques à Princeton associe l’informatique et la biologie pour développer des outils de base et des informations susceptibles d’avoir un impact considérable sur la santé et l’humanité.
« Les approches interdisciplinaires qui associent une science des données sophistiquée à une connaissance approfondie de la biologie sont essentielles pour déchiffrer les énigmes biomédicales nécessaires à la réalisation de la promesse de la médecine de précision » conclut Troyanskaya.
 L’acide désoxyribonucléique, communément appelé ADN, est une molécule complexe qui joue un rôle essentiel dans la biologie des organismes vivants. Il contient les instructions génétiques utilisées dans [...]
L’acide désoxyribonucléique, communément appelé ADN, est une molécule complexe qui joue un rôle essentiel dans la biologie des organismes vivants. Il contient les instructions génétiques utilisées dans [...]