
Une technique de microscopie révolutionnaire, dite cryoélectronique, permet pour la toute première fois d’observer des atomes individuels avec des détails sans précédent. Selon les chercheurs, cela « ouvre les portes d’un tout nouvel univers ».
Cette technique révolutionnaire pour l’imagerie des molécules est connue sous le nom de microscopie cryoélectronique (dite cryo-ME), qui correspond à une technique particulière de préparation d’échantillons biologiques utilisée en microscopie électronique en transmission. Cette technique permet de réduire les dommages d’irradiation causés par le faisceau d’électrons et permet également de préserver la morphologie et la structure des échantillons.
S’affranchissant de la présence de colorant ou de fixateur chimique, la technique consiste à congeler très rapidement des échantillons biologiques sous forme hydratée dans de l’éthane liquide, de manière à les figer dans leur état natif dans une glace amorphe (c’est-à-dire non cristalline). Avec le développement continu de nouvelles générations de microscopes électroniques et de systèmes d’acquisition des données (notamment le développement de caméras à détection directe d’électrons), la cryo-ME permet désormais dans certains cas d’obtenir des résultats similaires, voire supérieurs, à ceux de la cristallographie aux rayons X pour la résolution de structures tridimensionnelles (3D) d’objets biologiques.
C’est cette technique qui a permis de produire les images les plus nettes à ce jour, et, pour la toute première fois, qui a permis de discerner des atomes individuels dans une protéine :

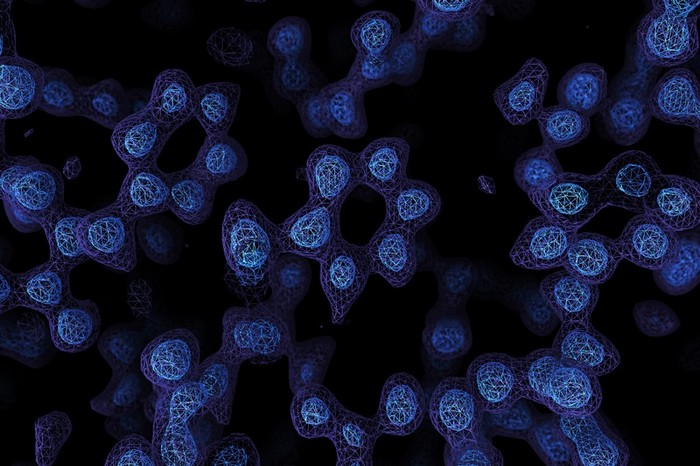 Une carte cryo-ME de la protéine apoferritine. Crédits : Paul Emsley/MRC Laboratory of Molecular Biology
Une carte cryo-ME de la protéine apoferritine. Crédits : Paul Emsley/MRC Laboratory of Molecular BiologyEn atteignant une résolution atomique à l’aide de la microscopie électronique cryogénique, les chercheurs seront en mesure de comprendre, avec des détails sans précédent, le fonctionnement des protéines qui ne peuvent pas être facilement examinées par d’autres techniques d’imagerie, telles que la cristallographie aux rayons X (une technique d’analyse fondée sur la diffraction des rayons X par la matière, particulièrement quand celle-ci est cristalline). La diffraction des rayons X est une diffusion élastique, c’est-à-dire sans perte d’énergie, des photons (longueurs d’onde inchangées), qui donne lieu à des interférences d’autant plus marquées que la matière est ordonnée. Pour les matériaux non cristallins, on parle plutôt de diffusion.
Cette percée, signalée par deux laboratoires à la fin du mois dernier, « cimente la position de la microscopie cryo-ME en tant qu’outil dominant pour cartographier les formes 3D des protéines », affirment les scientifiques. En fin de compte, ces structures aideront les chercheurs à comprendre le fonctionnement des protéines dans le domaine de la santé et dans diverses maladies, et conduiront à de meilleurs traitements, avec moins d’effets secondaires.
« C’est vraiment un pas en avant, c’est sûr. Il n’y a vraiment plus rien à franchir dans ce domaine. Cela a été la dernière barrière de résolution », explique Holger Stark, biochimiste et microscopiste électronique à l’Institut Max Planck de chimie biophysique à Göttingen, en Allemagne, qui a dirigé l’une des études (¹). Une autre étude (²) a été dirigée par Sjors Scheres et Radu Aricescu, biologistes structurels au laboratoire de biologie moléculaire du Medical Research Council (MRC-LMB) à Cambridge, au Royaume-Uni. Les deux études ont été publiées sur le serveur de préimpression bioRxiv le 22 mai.
« La véritable ‘résolution atomique’ est un réel jalon », affirme John Rubinstein, biologiste des structures à l’Université de Toronto au Canada. « Obtenir des structures à résolution atomique de nombreuses protéines sera toujours une tâche ardue en raison d’autres défis, tels que la flexibilité d’une protéine. Mais ces préimpressions montrent où l’on peut arriver si ces autres limitations peuvent être résolues », explique-t-il.
Dépasser les limites
La cryo-ME est une technique datant d’il y a plusieurs décennies qui détermine la forme des échantillons gelés par flash en leur projetant des électrons et en enregistrant les images résultantes. Les progrès de cette technologie de détection des électrons à ricochets et des logiciels d’analyse d’images ont catalysé une « révolution de la résolution », qui a débuté vers 2013. Cela a conduit à des visualisations des structures protéiques plus nettes que jamais (et presque aussi bonnes que celles obtenues par cristallographie aux rayons X, une technique plus ancienne qui déduit les structures des modèles de diffraction faits par les cristaux de protéines lorsqu’ils sont bombardés de rayons X).
Les avancées matérielles et logicielles ultérieures ont permis d’améliorer davantage la résolution des structures cryo-ME. Mais les scientifiques ont dû largement s’appuyer sur la cristallographie aux rayons X pour obtenir des structures à résolution atomique. Cependant, les chercheurs peuvent passer des mois, voire des années, à cristalliser une protéine, et de nombreuses protéines médicalement importantes ne formeront pas de cristaux utilisables. En revanche, la cryo-ME nécessite uniquement que la protéine soit dans une solution purifiée.
Les cartes à résolution atomique sont suffisamment précises pour discerner sans ambiguïté la position des atomes individuels dans une protéine, à une résolution d’environ 1,2 ångström. Ces structures sont particulièrement utiles pour comprendre le fonctionnement des enzymes et utiliser ces informations pour identifier les médicaments qui peuvent bloquer leur activité. À noter qu’un ångström est une unité de longueur valant 0,1 nanomètre, soit 10−10 mètre (un dixième de milliardième de mètre) et ayant pour symbole Å. Il s’agit d’une unité fréquemment utilisée en physique atomique.
Pour pousser la cryo-ME à la résolution atomique, les deux équipes ont travaillé sur une protéine stockant le fer appelée apoferritine. En raison de sa stabilité rocheuse, la protéine est devenue un banc d’essai pour la cryo-ME : une structure de la protéine avec une résolution de 1,54 ångström, était le précédent record (³).

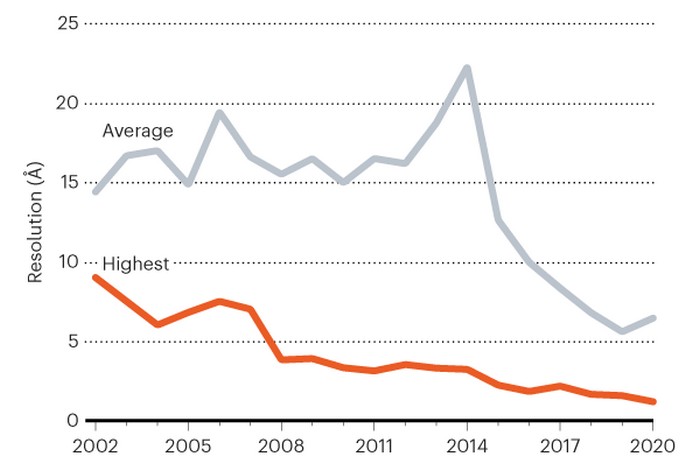 La technique d’imagerie des atomes connue sous le nom de microscopie cryoélectronique (dit cryo-ME) peut à présent dévoiler des caractéristiques au niveau atomique, soit d’environ 1,2 ångström. En gris, la moyenne. En rouge, la plus haute résolution. Crédits : Nature
La technique d’imagerie des atomes connue sous le nom de microscopie cryoélectronique (dit cryo-ME) peut à présent dévoiler des caractéristiques au niveau atomique, soit d’environ 1,2 ångström. En gris, la moyenne. En rouge, la plus haute résolution. Crédits : NatureMais les chercheurs ont alors utilisé les améliorations technologiques pour prendre des photos plus nettes encore de l’apoferritine : l’équipe de Stark a obtenu une structure de 1,25-ångström de la protéine, avec l’aide d’un instrument qui garantit que les électrons voyagent à des vitesses similaires avant de toucher un échantillon, améliorant la résolution des images résultantes.
Scheres, Aricescu et leur groupe ont utilisé une technologie différente pour tirer des électrons se déplaçant à des vitesses similaires : ils ont également bénéficié d’une technologie qui réduit le bruit généré par certains électrons sur l’échantillon de protéines, ainsi que d’une caméra de détection d’électrons plus sensible. « La structure de 1,2-ångström était si complète que nous pouvions déterminer les atomes d’hydrogène individuels, à la fois dans la protéine et dans les molécules d’eau environnantes », dit Scheres.
Stark estime que la fusion des technologies pourrait pousser les résolutions à environ 1 ångström, mais pas beaucoup plus loin : « Il est presque impossible d’atteindre un chiffre situé en dessous de 1 Å avec la cryo-EM », explique-t-il. En effet, obtenir une telle structure avec la technologie de pointe existante nécessiterait « plusieurs centaines d’années d’enregistrement de données et une quantité non réaliste de puissance de calcul et de capacités de stockage de données », estime l’équipe de recherche.
Ouvrir les portes d’un nouvel univers
Scheres et Aricescu ont également testé leurs améliorations sur une forme simplifiée d’une protéine appelée récepteur GABAA. Cette protéine se trouve dans la membrane des neurones et est une cible pour les anesthésiques généraux, les médicaments contre l’anxiété et de nombreux autres médicaments. L’année dernière, l’équipe d’Aricescu a utilisé la cryo-ME pour cartographier la protéine à 2,5 ångströms (4). Mais avec le nouveau kit, les chercheurs ont atteint une résolution de 1,7 ångström, avec une résolution encore meilleure dans certaines parties clés de la protéine : « C’était comme enlever des poussières qui s’étaient logées sur nos yeux », explique Aricescu. « Avec cette résolution, chaque demi-ångström ouvre la porte vers un univers entier », a ajouté Aricescu.
En effet, la structure a révélé des détails inédits dans la protéine, y compris des molécules d’eau dans la poche où se trouve un produit chimique appelé histamine : « C’est une mine d’or pour la conception de médicaments basés sur la structure », explique Aricescu, car elle montre comment un médicament pourrait déplacer les molécules d’eau, ce qui pourrait entraîner l’élaboration de médicaments avec moins d’effets secondaires. « Établir une carte à résolution atomique de GABAA, qui n’est pas aussi stable que l’apoferritine, serait un défi. Je ne pense pas que ce soit impossible, mais ce ne serait pas très pratique, en raison de la grande quantité de données qui devraient être collectées », explique Scheres. Mais d’autres améliorations, en particulier dans la manière dont les échantillons de protéines sont préparés, pourraient ouvrir la voie à des structures de résolution atomique de GABAA, ainsi que d’autres protéines biomédicalement importantes.
« Tout le monde est très excité et étonné par le niveau de performance vraiment stupéfiant démontré par les groupes MRC-LMB et Max Planck », déclare Radostin Danev, spécialiste de la cryo-ME à l’Université de Tokyo. Mais il convient que la préparation des échantillons est le défi majeur du domaine pour des protéines plus bancales : « Les performances de résolution inférieures à 1,5 Å, voire inférieures à 2 Å, resteront accessibles pendant un certain temps uniquement aux échantillons qui se comportent bien », ajoute Danev. « Les avancées technologiques sont susceptibles de consolider la position de la cryo-ME en tant qu’outil incontournable pour la plupart des études structurelles », explique Scheres. En effet, les sociétés pharmaceutiques qui convoitent les structures de résolution atomique pourraient être encore plus susceptibles de se tourner vers la cryo-ME à présent.
Cependant, Stark pense que la cristallographie aux rayons X conservera un certain attrait. Si une protéine peut être cristallisée, alors il est relativement efficace d’en générer des structures liées à des milliers de médicaments potentiels en peu de temps. Mais cela peut prendre des heures, voire des jours, pour générer suffisamment de données pour des structures cryo-ME à très haute résolution. « Il y a encore des avantages et des inconvénients pour chacune des techniques. Les gens ont publié de nombreux articles et critiques affirmant que les dernières avancées en cryo-ME signeraient l’arrêt de mort des rayons X. Mais j’en doute », ajoute Stark.
 L’électron est une particule élémentaire qui, avec les protons et les neutrons, constitue les atomes. C’est donc l’un des composants principaux de la matière baryonique. À ce titre, il revêt... [...]
L’électron est une particule élémentaire qui, avec les protons et les neutrons, constitue les atomes. C’est donc l’un des composants principaux de la matière baryonique. À ce titre, il revêt... [...]