
Depuis un peu plus d’un an maintenant, le monde entier est focalisé sur le virus SARS-CoV-2. Les scientifiques ont par ailleurs averti que d’autres virus sont susceptibles d’émerger dans les années à venir, ce qui peut paraître effrayant. Pourtant, sans le savoir, nous hébergeons nous-mêmes dans notre organisme des dizaines de milliers d’espèces virales : des chercheurs viennent d’en recenser 142 809 dans nos intestins, dont plus de la moitié étaient inconnues de la science auparavant.
Leurs travaux ont abouti à la création d’un nouveau catalogue de virus, nommé Gut Phage Database. Pour recenser les différentes espèces virales, l’équipe a analysé plus de 28 000 métagénomes individuels, issus de séquençage d’ADN d’échantillons de microbiome intestinal collectés dans 28 pays, ainsi que près de 2900 génomes de référence de bactéries intestinales cultivées. Les échantillons provenaient d’individus en bonne santé, qui ne présentaient aucune maladie spécifique.
Ces virus qui vivent dans l’intestin humain sont de type bactériophage ; ils sont capables d’infecter les bactéries et les archées. Tous les virus ne sont pas nocifs pour l’Homme. Les bactériophages jouent par exemple un rôle important pour notre santé, en régulant les bactéries qui se trouvent dans notre intestin. « Ils font partie intégrante de l’écosystème intestinal », explique le biochimiste Alexandre Almeida de l’Institut de bio-informatique du Laboratoire européen de biologie moléculaire (EMBL-EBI) et de l’Institut Wellcome Sanger.
Plus de 40 000 génomes viraux de haute qualité
Les virus sont les entités biologiques les plus nombreuses sur Terre avec une population estimée à 1031 particules ! Les bactériophages influencent profondément les communautés microbiennes en fonctionnant comme des vecteurs de transfert horizontal de gènes, en codant des fonctions utiles pour les espèces bactériennes hôtes et en promouvant des interactions co-évolutives dynamiques. Mais pendant longtemps, le phénomène était mal compris des scientifiques.
Ces dernières années, les avancées en matière d’analyse métagénomique ont permis d’obtenir de nouvelles informations fondamentales sur la diversité et les fonctions virales présentes dans le microbiome intestinal humain. Néanmoins, le nombre limité de métagénomes utilisés pour construire les premières bases de données (moins de 700) implique que la majorité de la diversité des phages intestinaux (les virus bactériens) restait non caractérisée et incomplète. La Gut Phage Database (GPD) vient combler ces lacunes : elle comprend plus de 40 000 génomes de phages de haute qualité.
« À notre connaissance, cet ensemble représente la collection la plus complète et la plus précise de génomes de phages intestinaux humains à ce jour », écrivent les auteurs de l’étude. Disposer aujourd’hui d’une base de données complète de génomes de phages de haute qualité ouvre la voie à une multitude d’analyses du virome intestinal humain (la composante virale du microbiote) à une résolution grandement améliorée. Pour Trevor Lawley, microbiologiste au Wellcome Sanger Institute, « la recherche sur les bactériophages connaît actuellement une renaissance ».
Pour commencer, ce catalogue a permis d’actualiser les connaissances des scientifiques concernant le comportement viral. Après avoir regroupé l’ensemble du protéome de la GPD en 202’192 grappes de protéines, les chercheurs ont constaté que les fonctions supérieures correspondaient aux protéines de liaison de l’ADN, aux intégrases, aux méthylases, aux peptidases et aux protéines ruban ; cependant, la majorité des protéines phagiques (47,46%) n’ont pas pu se voir attribuer une fonction.
Plus d’un tiers des virus ciblent plusieurs espèces de bactéries
La GPD a également permis d’attribuer un phage spécifique à des espèces hôtes bactériennes. Les chercheurs ont notamment vérifié s’il y avait une préférence pour l’infection phagique via 4 phylums bactériens intestinaux humains communs (Firmicutes, Bacteroidetes, Proteobacteria et Actinobacteriota) : ils ont ainsi détecté une prévalence significativement plus faible des phages dans Actinobacteriota, avec 59% d’isolats infectés contre au moins 70% pour les autres phylums.

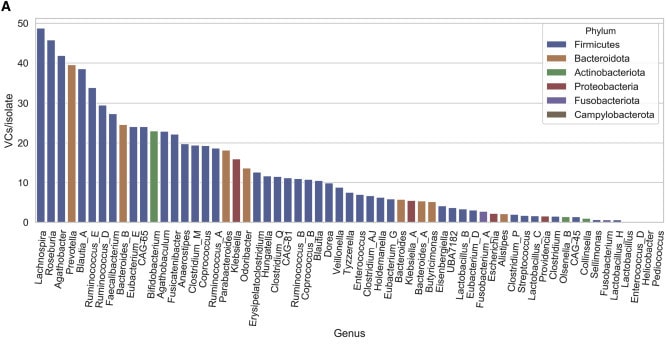 Les genres bactériens avec la plus grande diversité virale étaient Lachnospira, Roseburia, Agathobacter, Prevotella et Blautia A. D’autre part, la plus faible diversité virale était hébergée par Helicobacter et les bactéries lactiques Lactobacillus, Lactobacillus H, Enterococcus D et Pediococcus. © L. Camarillo-Guerrero et al.
Les genres bactériens avec la plus grande diversité virale étaient Lachnospira, Roseburia, Agathobacter, Prevotella et Blautia A. D’autre part, la plus faible diversité virale était hébergée par Helicobacter et les bactéries lactiques Lactobacillus, Lactobacillus H, Enterococcus D et Pediococcus. © L. Camarillo-Guerrero et al.Puis, les chercheurs ont évalué la diversité virale au sein de chaque phylum. Cette analyse a révélé que les Firmicutes abritent une diversité virale significativement plus élevée que les autres phylums. L’analyse au niveau du genre bactérien dans tous les phylums a révélé que Lachnospira, Roseburia, Agathobacter, Prevotella et Blautia A contiennent le plus grand nombre de grappes virales par isolat ; en revanche, la plus faible diversité virale par isolat a été détectée parmi Helicobacter et les bactéries lactiques Lactobacillus H, Lactobacillus, Enterococcus D et Pediococcus. Ces résultats mettent en évidence la large distribution de l’abondance et de la prévalence des phages parmi les bactéries intestinales humaines, même au sein d’un même phylum.


Les chercheurs ont découvert par ailleurs que plus d’un tiers (36%) des grappes virales ne sont pas limitées à infecter une seule espèce, ce qui crée des réseaux de flux de gènes à travers des espèces bactériennes phylogénétiquement distinctes. De plus, ils ont identifié 280 grappes virales réparties dans le monde, y compris un clade nouvellement identifié, très répandu, appelé Gubaphage ; il semble être le deuxième clade de virus le plus répandu dans l’intestin humain, après le groupe que l’on nomme p-crAssphage.

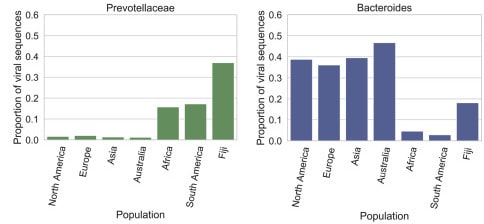 La proportion de séquences virales qui ciblent les hôtes Prevotellaceae dans les sociétés traditionnelles est plus élevée que celle des populations industrialisées. À l’inverse, les hôtes Bacteroides sont plus courants dans les populations industrialisées que dans les sociétés traditionnelles. Ce résultat suggère que la composition du phageome intestinal à l’échelle mondiale est déterminée par la composition bactérienne. © L. Camarillo-Guerrero et al.
La proportion de séquences virales qui ciblent les hôtes Prevotellaceae dans les sociétés traditionnelles est plus élevée que celle des populations industrialisées. À l’inverse, les hôtes Bacteroides sont plus courants dans les populations industrialisées que dans les sociétés traditionnelles. Ce résultat suggère que la composition du phageome intestinal à l’échelle mondiale est déterminée par la composition bactérienne. © L. Camarillo-Guerrero et al.Ils ont observé une séparation claire entre les phageomes nord-américains, européens et asiatiques et les phageomes africains et sud-américains ; ces modèles de phageome sont associés à des différences importantes dans les modes de vie humains. En effet, les bactéries Prevotellaceae sont plus abondantes et prévalentes chez les individus ayant un mode de vie rural/traditionnel, tandis que les Bacteroides sont plus abondantes et prévalentes chez les individus ayant un mode de vie urbain/occidental.
Ce n’est là qu’un aperçu des informations fournies par la GPD nouvellement établie et les chercheurs se réjouissent des découvertes à venir, grâce à cette nouvelle base de connaissances. « Ce catalogue de grande qualité et à grande échelle de génomes de phages améliorera les futures études sur les viromes et permettra une analyse écologique et évolutive des bactériophages intestinaux humains », concluent-ils.
