
Les protéines sont des molécules essentielles au vivant, et leur fonction dépend en grande partie de la façon dont la chaîne d’acides aminés qui les compose « se replie » dans l’espace, après avoir été synthétisée par la cellule. Ce repliement des protéines est l’un des problèmes les plus épineux de la biologie, alimentant, depuis plus de cinquante ans, de nombreux travaux. Récemment, des chercheurs, en combinant l’IA avec des techniques expérimentales et informatiques, ont mis en évidence l’architecture du complexe de pores nucléaires humains de la façon la plus détaillée jusqu’ici. Il s’agit du plus grand complexe protéique de la cellule, d’environ 1000 protéines qui perforent l’enveloppe nucléaire et assurent le transport des instructions de l’ADN vers le reste de la cellule. Ce travail illustre comment la modélisation basée sur l’IA peut être intégrée à la biologie structurale in situ pour comprendre les principes de construction et de conception des grands assemblages moléculaires. Il définit une perspective révolutionnaire dans la recherche médicale.
Le complexe de pores nucléaires humain (NPC) est un véritable géant moléculaire. Ces canaux sont essentiels à la vie. Intégrés dans la barrière à double membrane, ou enveloppe nucléaire — qui sépare le noyau et son ADN du reste de la cellule —, ils contrôlent étroitement quelles macromolécules entrent ou sortent du noyau. Ces activités consistent notamment à permettre aux protéines vitales d’entrer dans le noyau, à bloquer les virus et à faire passer les ARN messagers du noyau au cytoplasme, où ils sont traduits en protéines. Le système de transport nucléaire joue également un rôle dans plusieurs maladies, notamment les troubles neurodégénératifs, le cancer et les infections virales.
Aux yeux des biologistes structuraux, le complexe de pores nucléaires humain est un puzzle 3D passionnant, avec environ 30 protéines différentes, présentes chacune en plusieurs exemplaires. Cela équivaut à environ 1000 pièces de puzzle, qui forment un noyau rond entouré de pièces flexibles. Jusqu’à présent, les modèles les plus précis ne couvraient que 46% de la structure.
Récemment, le groupe Kosinski de l’EMBL de Hambourg et du Centre de biologie des systèmes structurels (CSSB), des laboratoires Beck et Hummer de l’Institut Max Planck de biophysique, ont créé le modèle le plus complet à ce jour, en combinant le programme de prédiction de la structure des protéines AlphaFold2 (créé par la société DeepMind de Google) avec des techniques telles que la tomographie cryoélectronique, la cryo-EM à particule unique et la modélisation intégrative. Leur modèle couvre plus de 90% de la structure. Il a été publié dans la revue Science.
Expérimentation et intelligence artificielle, un duo gagnant
Pour atteindre ce résultat, jugé comme un triomphe de la biologie structurale expérimentale, les scientifiques, riches de deux décennies de recherches dans le domaine, ont combiné plusieurs méthodes expérimentales et informatiques. Cela leur a permis de visualiser le PNC à différentes échelles et niveaux de détails.
Dans un premier temps, pour modéliser la silhouette globale du complexe protéique, les chercheurs ont utilisé la cryo-tomographie électronique. Grâce à cette technique, ils ont pu l’observer dans son environnement cellulaire, plutôt qu’isolé. Puis, AlphaFold2, un programme basé sur l’intelligence artificielle prédisant les structures des protéines, a révélé plus de détails sur les éléments constitutifs des protéines individuelles.
Agnieszka Obarska-Kosińska, postdoc qui a effectué la modélisation moléculaire, déclare dans un communiqué : « AlphaFold2 a été un moment décisif pour nous. Avant, nous ne connaissions pas la structure de nombreuses protéines au sein du complexe. Vous ne pouvez pas assembler un puzzle si vous ne savez pas à quoi ressemblent les pièces. Mais AlphaFold2, combiné à d’autres approches, nous a permis de prédire ces formes ».
Dans un second temps, l’équipe a utilisé ColabFold, une version d’AlphaFold2 modifiée par la communauté scientifique pour modéliser les interactions entre protéines. Cette IA leur a permis de visualiser comment les différentes pièces du puzzle se combinent pour former des sous-complexes plus petits, et comment ces sous-complexes sont ensuite assemblés pour former le complexe de pores nucléaires.

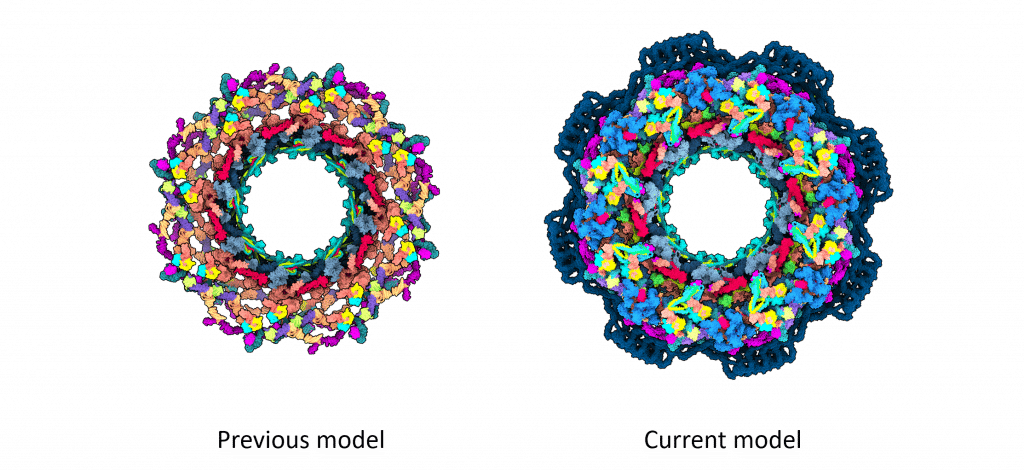 Comparaison des modèles de complexes de pores nucléaires humains obtenus avant (à gauche) et après l’utilisation d’AlphaFold2 combiné à la cryo-ET (à droite). © Mosalaganti et al., 2022
Comparaison des modèles de complexes de pores nucléaires humains obtenus avant (à gauche) et après l’utilisation d’AlphaFold2 combiné à la cryo-ET (à droite). © Mosalaganti et al., 2022Enfin, les chercheurs ont assemblé toutes les pièces à l’aide du logiciel Assembline, précédemment développé par le groupe Kosinski, et l’ont validé par rapport à des données expérimentales.
Jan Kosinski, chef du groupe EMBL, et qui a co-dirigé l’étude, explique : « C’est comme quand on démonte et remonte un appareil électronique. Il restera toujours quelques vis, et vous ne savez tout simplement pas où elles sont censées se trouver. Nous avons finalement réussi à équiper la plupart d’entre eux, et maintenant, nous savons exactement où ils se trouvent, ce qu’ils font et comment ».
Le modèle résultant est si complet et détaillé qu’il a permis aux chercheurs de créer des simulations moléculaires résolues dans le temps, expliquant comment les protéines du complexe et la membrane nucléaire interagissent pour créer un pore stable. Ils ont également pu appréhender la manière dont il répond aux signaux mécaniques.


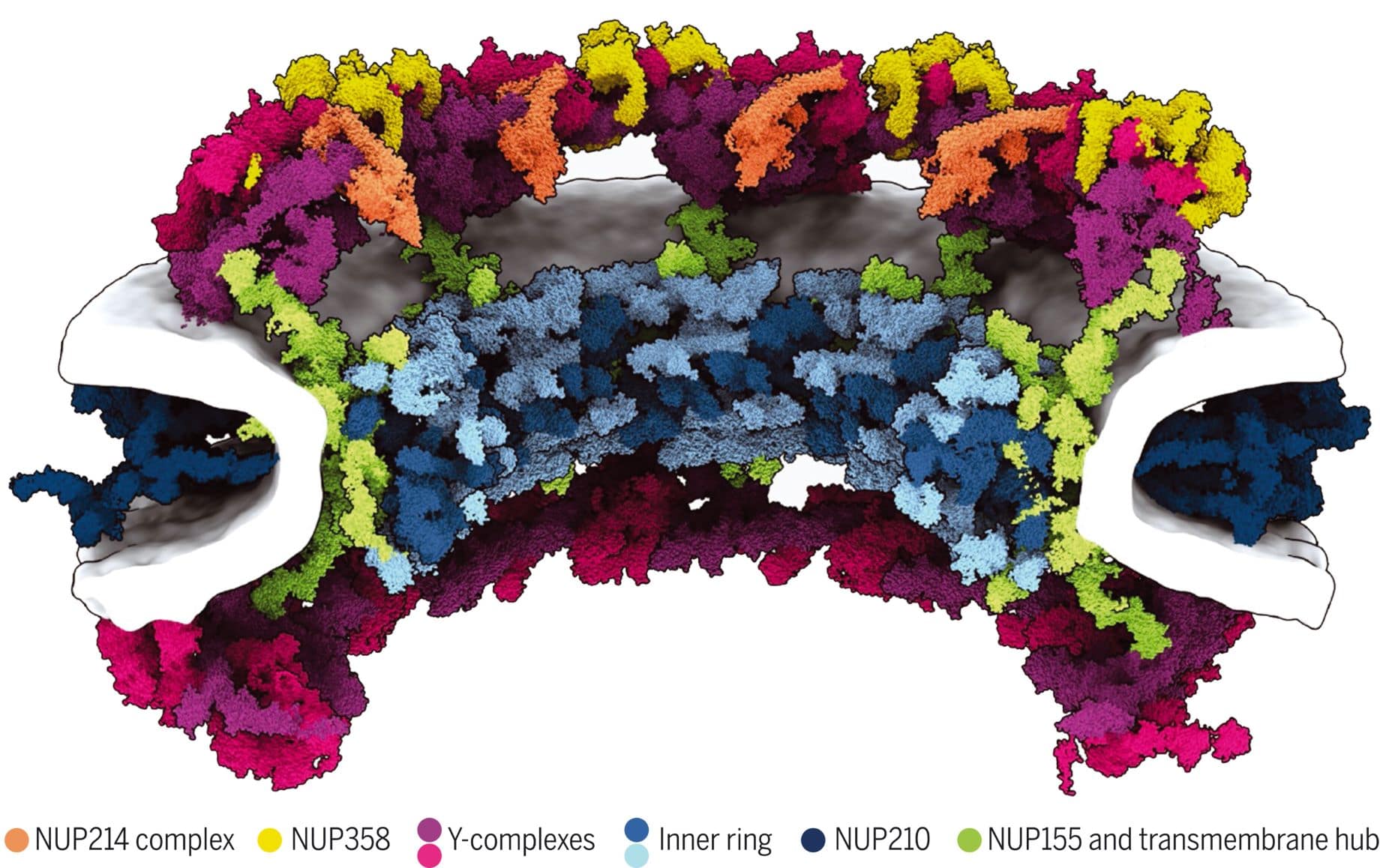 Vue en coupe de l’architecture d’échafaudage complexe de pores nucléaires humains, à l’état resserré. Les modèles haute résolution sont codés par couleur en fonction de l’appartenance au sous-complexe. L’enveloppe nucléaire est représentée par une surface grise. © Mosalaganti et al., 2022
Vue en coupe de l’architecture d’échafaudage complexe de pores nucléaires humains, à l’état resserré. Les modèles haute résolution sont codés par couleur en fonction de l’appartenance au sous-complexe. L’enveloppe nucléaire est représentée par une surface grise. © Mosalaganti et al., 2022Une découverte essentielle pour la recherche médicale
Ce travail représente un grand pas en avant dans la compréhension de l’architecture des pores nucléaires. En effet, comme mentionné précédemment, outre la coordination du transport, ces complexes protéiques organisent des processus nucléaires et cytoplasmiques essentiels tels que la transcription et la maturation de l’ARNm. Ces divers rôles en font « un hotspot », comme le précise le rédacteur en chef de Science dans un papier associé à l’étude, pour les mutations associées à la maladie et les interactions hôte-pathogène. Par conséquent, ils sont essentiels pour la thérapie génique, les vaccins de type ARNm, CRISPR et d’autres thérapies géniques.
Le groupe Kosinski orientera ses travaux futurs vers le développement de méthodes automatiques d’intégration de données structurelles et microscopiques à l’aide d’AlphaFold2 et de son propre logiciel Assembline. Ils prévoient d’appliquer ces approches à l’étude des processus moléculaires à l’origine des infections virales.
Les Drs Gerhard Hummer et Martin Beck, de l’Institut Max Planck de biophysique, soulignent : « Ce travail illustre comment, à l’avenir, la biologie structurale embrassera la biologie cellulaire pour créer des modèles atomiques d’assemblages toujours plus grands de molécules qui remplissent différentes fonctions dans différentes parties de la cellule ».
Cette présente étude fait partie d’un corpus de cinq articles publiés simultanément dans la revue Science, révélant une image quasi atomique de l’énorme complexe de pores nucléaires humain. Ces résultats permettront aux futures recherches de sonder davantage les nombreuses fonctions des complexes protéiques.
