
En utilisant l’outil d’IA de prédiction de protéines AlphaFold développé par Google DeepMind, des chercheurs ont identifié des centaines de milliers de nouvelles molécules psychédéliques pouvant potentiellement servir d’antidépresseurs. Bien que l’outil ait suscité le scepticisme quant à sa fiabilité, la nouvelle étude suggère que les structures prédites pourraient être tout aussi utiles pour la découverte de nouveaux médicaments que celles dérivées expérimentalement.
La technique conventionnelle pour le développement de médicaments consiste à modéliser des centaines de millions de structures moléculaires (voire des milliards) et de déterminer comment elles réagissent avec une protéine cible. Appelé « amarrage protéine-ligand », ce processus permet d’identifier les molécules capables de modifier de manière optimale l’activité de la protéine de sorte à induire l’effet thérapeutique recherché.
Les techniques d’amarrage protéine-ligand s’appuient principalement sur des structures protéiques expérimentales, obtenues par exemple par cristallographie à rayons X (une technique d’analyse fondée sur la diffraction des rayons X par la matière) ou par cryomicroscopie électronique.
Cependant, pour de nombreuses cibles médicamenteuses, les données sur de telles structures expérimentales ne sont pas disponibles, sans compter que les étapes d’amarrage protéine-ligand peuvent prendre plusieurs mois voire des années. Les outils de prédiction de structures protéiques tels qu’AlphaFold ont été développés dans le but de combler ces lacunes et d’accélérer les processus de développement de médicaments.
La base de données publique d’AlphaFold contient des prédictions de structure pour presque toutes les protéines connues. L’outil présente notamment les structures protéiques du protéome humain et des protéomes de 47 autres organismes clés, couvrant plus de 200 millions de protéines et presque toutes les cibles protéiques thérapeutiques potentielles.
Alors que certains experts estiment que l’outil constitue une véritable révolution pour la recherche biomédicale, d’autres en revanche sont sceptiques et remettent en question sa fiabilité. Ces derniers pointent notamment du doigt la capacité des prédictions d’AlphaFold à remplacer les modèles de structure expérimentaux. Selon Brian Shoichet, chimiste pharmaceutique à l’Université de Californie à San Francisco, « il y a beaucoup de battage médiatique. Quand quelqu’un affirme que tel ou tel va révolutionner la découverte de médicaments, cela justifie un certain scepticisme ».
D’un autre côté, plus d’une dizaine de recherches ont suggéré que les prédictions d’AlphaFold étaient moins utiles que les structures protéiques obtenues avec des méthodes expérimentales, lorsqu’elles sont utilisées pour identifier des médicaments potentiels. Plus important encore, l’outil serait incapable de distinguer les nouveaux médicaments de ceux déjà connus. Shoichet et ses collègues se sont alors demandé si les petites différences des structures expérimentales par rapport à celles prédites pourraient amener l’outil d’IA à manquer certains composés pouvant se lier aux protéines.
D’autre part, le modèle pourrait aussi en identifier des différents, mais qui ne seraient pas moins prometteurs. Si tel était le cas, les structures prédites pourraient en fin de compte être aussi utiles que celles expérimentales. C’est ce que les experts ont cherché à déterminer dans cette nouvelle étude, prépubliée sur la plateforme biorXiv.
Des molécules différentes, mais avec des taux de réussite identiques
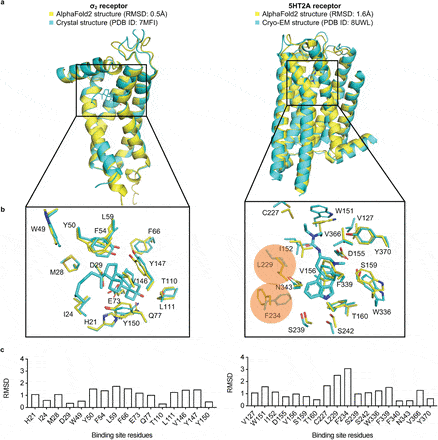
Afin d’explorer leur hypothèse, les chercheurs ont sélectionné les structures expérimentales de deux protéines pour tester des centaines de millions de médicaments potentiels (ligands). L’une des protéines est le récepteur de la sérotonine (récepteur 5-HT2A), dont la structure a été déterminée par cryomicroscopie électronique. La seconde est le récepteur sigma-2, se liant avec la testostérone, la progestérone et le cholestérol.
Le même processus a été effectué avec des modèles de protéines issus de la base de données AlphaFold. Des centaines de composés considérés prometteurs et modélisés par le biais des deux processus ont ensuite été sélectionnés pour être synthétisés et pour l’analyse de leur activité en laboratoire.

Les chercheurs ont constaté que les criblages avec les structures prédites et expérimentales ont donné des ligands complètement différents. « Il n’y avait pas deux molécules identiques », explique Shoichet. « Ils ne se ressemblaient même pas un peu », ajoute-t-il. Cependant, de manière surprenante, les taux de réussite (c’est-à-dire leur capacité à modifier l’activité des protéines cibles) des composés issus des deux groupes étaient presque identiques, malgré leurs différences.
De plus, les ligands les plus puissants pour le récepteur 5-HT2A ont été identifiés par le biais de l’amarrage avec les structures prédites par AlphaFold, et non avec celles expérimentales. Les psychédéliques agissant en partie par le biais de cette voie, cela suggère qu’il est possible d’identifier des versions agissant comme des antidépresseurs, mais qui ne seraient pas hallucinogènes.
Un scepticisme qui subsiste
Toutefois, le scepticisme subsiste, car ces performances pourraient ne pas être reproductibles pour d’autres cibles médicamenteuses. Une étude distincte a notamment montré que dans environ 10 % des cas, les structures qu’AlphaFold considère comme très précises sont considérablement différentes de celles générées expérimentalement. Cela suggère que les chercheurs utilisant l’outil devraient tout de même s’appuyer sur les modèles expérimentaux plus détaillés pour optimiser les propriétés d’un médicament candidat. En d’autres termes, l’outil peut fournir de précieuses pistes de recherche, mais ne serait pas encore en mesure de supplanter les structures expérimentales.
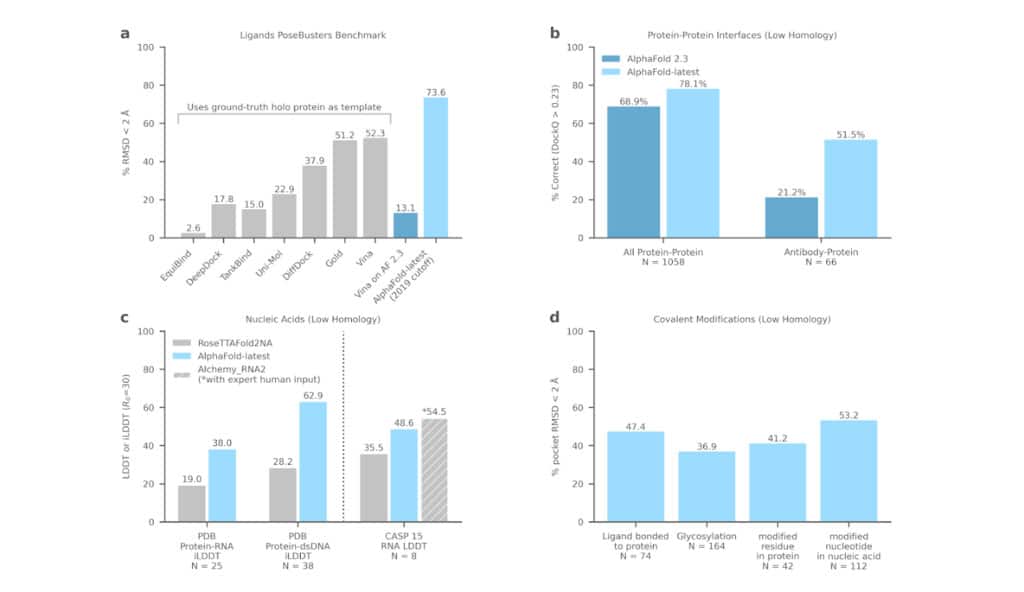
Toutefois, dans un tiers des cas, AlphaFold pourrait prédire une structure dont le potentiel pourrait permettre de relancer un projet, selon Shoichet. « Par rapport à l’acquisition d’une nouvelle structure, vous pourriez faire avancer le projet de quelques années et c’est énorme », déclare-t-il. Par ailleurs, Google DeepMind a récemment annoncé avoir apporté des améliorations à l’outil. Sa dernière version peut désormais générer des prédictions pour presque toutes les molécules de la Protein Data Bank (PDB), une base de données mondiale sur la structure tridimensionnelle de macromolécules biologiques (protéines et acides nucléiques). Cela apporterait un gain de précision considérable à AlphaFold. La prochaine amélioration prévue ciblera la prédiction de la structure des protéines lorsqu’elles sont liées à des médicaments et à d’autres molécules en interaction.