
Un premier séquençage du génome humain, référencé GRCh38, a été achevé en 2003. Depuis, les progrès technologiques ont permis de peaufiner cet énorme travail, faisant du génome humain le génome de vertébré le plus complet jamais séquencé. Restaient cependant quelques lacunes à combler, notamment au niveau des chromosomes. Aujourd’hui, des chercheurs annoncent avoir réalisé le séquençage complet, sans aucune lacune, d’un chromosome X humain.
La séquence a été réalisée de bout en bout, autrement dit, de télomère à télomère (les régions se trouvant aux extrémités d’un chromosome). Aucun chromosome n’avait jamais été séquencé dans sa totalité jusqu’à présent. Pour ce faire, les généticiens ont exploité une technique, appelée séquençage par nanopores, qui consiste à déterminer l’ordre dans lequel les nucléotides sont disposés sur un fragment d’ADN donné et surtout, qui permet de réaliser des lectures ultra-longues de brins d’ADN.
Une combinaison de plusieurs techniques de séquençage
Les autres techniques de séquençage, au contraire, ne permettent que de lire de courtes sections d’ADN ; de ce fait, les généticiens doivent ensuite effectuer un laborieux travail de reconstitution, à la manière d’un puzzle, ce qui est potentiellement source d’erreurs. En effet, les différentes « pièces » à assembler sont très similaires et il est parfois difficile de savoir si elles sont placées dans le bon ordre et surtout, de s’assurer du nombre de répétitions dans la séquence. C’est pourquoi plusieurs lacunes subsistaient…
Les chercheurs ont rapidement découvert que ces données manquantes contenaient pourtant des informations essentielles : « Nous commençons à constater que certaines de ces régions où il y avait des lacunes […] sont en fait parmi les plus riches pour la variation des populations humaines, donc nous avons manqué beaucoup d’informations qui pourraient être importantes pour comprendre la biologie humaine », a déclaré Karen Miga, biologiste à l’Institut de génomique de Santa Cruz (Californie) et auteure principale de l’étude.
C’est pourquoi les généticiens se sont tournés vers le séquençage par nanopores. Le principe est d’utiliser de minuscules trous – d’un diamètre de l’ordre du nanomètre – fixés à une membrane électriquement résistante. Du courant est appliqué à la membrane, et ce courant électrique traverse ainsi le nanopore. Lorsque du matériel génétique est introduit dans ce dernier, le courant est modifié ; la quantité de courant varie avec la nature de ce qui obstrue le trou : une base A, T, G ou C, ou bien une section incluant plusieurs de ces bases nucléiques. Les variations de courant sont ainsi traduites en séquence génétique.
Non seulement cette technique permet des lectures de séquençage suffisamment longues, mais elle ne nécessite pas d’amplification d’ADN, qui peut également générer de nombreuses erreurs. Pour atteindre leur objectif, les chercheurs ont travaillé sur de l’ADN issu d’une môle hydatiforme (appelée aussi « grossesse môlaire ») — une tumeur bénigne rare se développant au cours de la grossesse. Cette lignée cellulaire spécifique, nommée CHM13, a déjà été utilisée par le passé pour combler les lacunes du génome humain (les génomes de ces grossesses sont uniformément homozygotes pour un ensemble d’allèles).

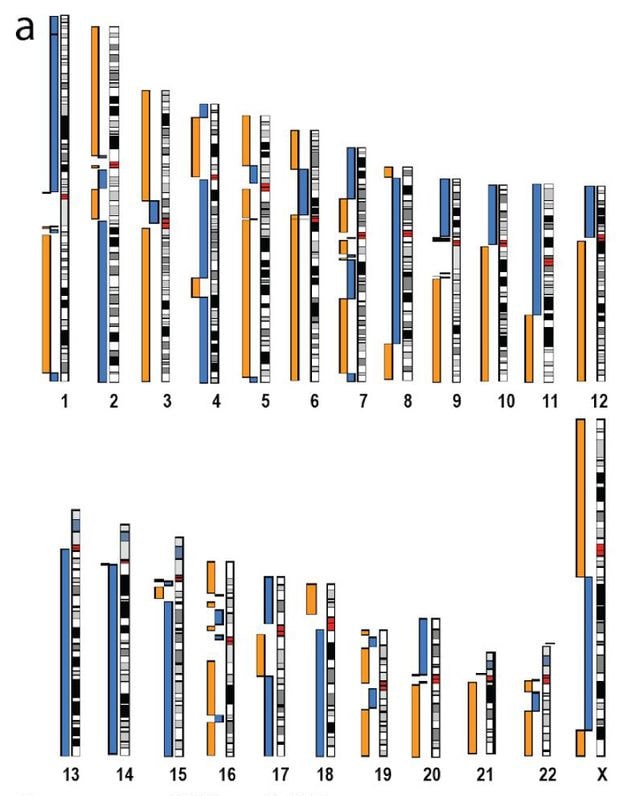 Assemblage du génome CHM13 en utilisant le séquençage par nanopores combiné au séquençage PacBio. Plusieurs chromosomes ne sont brisés que dans les régions centromériques (chr10, chr12, chr18, etc.). Crédits : K. Miga et al.
Assemblage du génome CHM13 en utilisant le séquençage par nanopores combiné au séquençage PacBio. Plusieurs chromosomes ne sont brisés que dans les régions centromériques (chr10, chr12, chr18, etc.). Crédits : K. Miga et al.Pour s’assurer d’obtenir un résultat aussi précis que possible, Miga et son équipe ont ensuite combiné leur méthode avec d’autres technologies de séquençage : Illumina et PacBio. Malgré tout, il restait encore des lacunes, notamment au niveau du centromère, la zone de jonction entre les deux chromatides.
Décrypter le génome pour mieux comprendre les maladies
Or, cette région particulièrement complexe est cruciale pour la mitose (l’étape où chaque chromosome de la cellule mère est dupliqué, puis réparti de façon égale dans chacune des deux cellules filles). Dans le chromosome X – l’un des deux chromosomes sexuels – c’est une région hautement répétitive, couvrant 3,1 millions de paires de bases d’ADN !

Les chercheurs ont pu résoudre cette structure notoirement délicate en recherchant de légères variations dans les répétitions. Ces variations leur ont permis de relier les longues lectures d’ADN pour reconstituer une séquence complète du centromère. Cette approche rigoureuse a finalement permis à l’équipe de combler les 29 lacunes du séquençage actuel du chromosome X. Un exploit encore difficile à croire pour Karen Miga : « Pour moi, l’idée que nous pouvons créer une répétition de 3 millions de paires de bases est tout simplement hallucinante ».
Sur le même sujet : Des chercheurs créent le premier génome autoréplicatif en laboratoire
Elle ajoute que le séquençage réalisé dans cette étude ouvre la voie au séquençage d’autres régions couvrant des millions de bases jugées jusqu’ici intraitables. Cette étude a également révélé une nouvelle séquence provenant des régions pseudo-autosomiques (des portions des chromosomes sexuels) et des familles de gènes ampliconiques du cancer du testicule (CT-X et GAGE) ; ces nouvelles séquences seront intégrées dans les futures versions du génome humain de référence.
Il s’agit d’une avancée majeure dans le projet de cartographie complète du génome humain : « Nos résultats démontrent que le séquençage de l’ensemble du génome humain est maintenant à portée de main et les données présentées ici permettront de poursuivre les efforts pour achever les chromosomes humains restants », se réjouit l’équipe. « L’achèvement de l’intégralité du génome humain devrait contribuer à notre compréhension de la fonction chromosomique et de la maladie humaine ».
