
Alors que de nombreux pays luttent toujours contre la seconde vague de la pandémie, plusieurs vaccins expérimentaux sont entrés ou viennent de terminer leur dernière phase d’essais cliniques. La prochaine étape pour les entreprises pharmaceutiques est de demander une autorisation pour une utilisation d’urgence. Cependant, l’accord d’une telle autorisation permet aux participants des essais placés initialement dans le groupe placebo de demander la vaccination. Or, pour beaucoup de scientifiques, cela pourrait poser un problème, car les entreprises risqueraient alors de ne pas disposer de suffisamment de données à long terme pour garantir la sécurité et l’innocuité des vaccins.
Suite à la publication des premières données des essais de phase III le 9 novembre, les fabricants de vaccins Pfizer et BioNTech ont demandé l’autorisation réglementaire de déployer leur vaccin selon les règles d’utilisation d’urgence. Le développeur d’un autre vaccin de premier plan, Moderna, devrait faire de même d’ici quelques semaines.
Une fois qu’un vaccin a reçu l’approbation d’urgence, il est demandé au fabricant de proposer l’immunisation aux participants à l’essai qui ont reçu un placebo. Mais si trop de personnes rejoignent le groupe vaccinal, les entreprises risquent de ne pas disposer de suffisamment de données pour établir des résultats à long terme, tels que la sécurité, la durée de la protection vaccinale et déterminer si le vaccin prévient l’infection ou simplement la maladie.
« C’est un vrai dilemme dans le développement de vaccins », déclare Klaus Stöhr, qui dirigeait auparavant la conception des vaccins chez la société pharmaceutique Novartis à Cambridge. Pourtant, Stöhr pense que le vaccin devrait recevoir une autorisation d’utilisation d’urgence, car son efficacité a été établie et le besoin est urgent.
Le dilemme posé par les autorisations d’urgence
Une telle compétition entre un essai clinique pour un vaccin et son utilisation d’urgence est nouvelle pour le développement de vaccins. Ce mois-ci seulement, l’Organisation mondiale de la santé a approuvé la toute première utilisation d’urgence pour une vaccination encore en phase de test, contre un type de poliovirus qui se propage dans l’hémisphère sud. Mais les essais de phase III pour ce vaccin n’ont pas encore commencé.
Pfizer, basée à New York, et BioNTech, basée à Mayence, en Allemagne, ont soumis le 20 novembre une demande d’autorisation d’utilisation d’urgence (EUA) auprès de la Food and Drug Administration (FDA) des États-Unis. En vertu des règles de la FDA pour les vaccins contre la COVID-19, les entreprises peuvent demander une EUA lorsque la moitié des participants à l’essai (la moitié des 43’000 personnes dans le cas de Pfizer) ont été suivis pendant deux mois après leur dernière dose. Pfizer et BioNTech remplissent déjà ce critère.
L’entreprise Moderna, basée à Cambridge, affirme qu’elle prévoit de franchir cette étape pour son essai de 30’000 participants bientôt, et qu’elle demandera un EUA dans les semaines à venir. La FDA a annoncé que son comité consultatif sur les vaccins se réunira le 10 décembre. Le comité évaluera les données des entreprises et décidera si les vaccins sont suffisamment sûrs et efficaces pour un usage restreint.

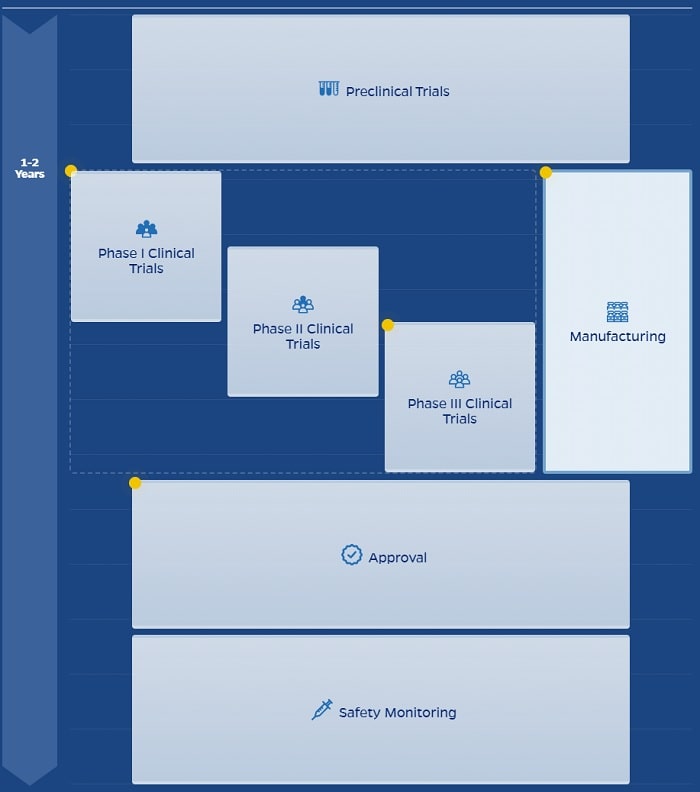 Chronologie du développement accéléré d’un vaccin. Les points jaunes indiquent les étapes qui peuvent être modifiées pour un développement plus rapide. Comme la fusion des phases I et II, ou II et III. Crédits : JHU
Chronologie du développement accéléré d’un vaccin. Les points jaunes indiquent les étapes qui peuvent être modifiées pour un développement plus rapide. Comme la fusion des phases I et II, ou II et III. Crédits : JHUDe nombreux chercheurs s’attendent à ce que les autorisations soient accordées. Une fois qu’un vaccin est approuvé, un comité des Centres pour le contrôle et la prévention des maladies (CDC) déterminera quels groupes devraient être les premiers en ligne pour la vaccination. Le panel examine les groupes à haut risque, tels que les personnes âgées, les personnes atteintes de maladies telles que le diabète qui les rendent plus sensibles à la COVID-19 et les soignants.
L’utilisation précoce des vaccins dans des groupes à haut risque sauvera presque certainement de nombreuses vies, déclare Jerome Kim, directeur général de l’Institut international des vaccins à Séoul. Cependant, les vaccins n’ont été testés que depuis quelques mois, il est donc trop tôt pour savoir pendant combien de temps ils seront efficaces.
Échanges entre groupes : un risque de biais dans les données
Les participants à l’essai ne savent généralement pas s’ils ont reçu le vaccin ou un placebo. Mais une fois qu’un vaccin a démontré son efficacité, il devient plus difficile de demander aux participants de rester dans le groupe placebo sans protection, déclare Paul Offit, chercheur en immunologie au Children’s Hospital de Philadelphie. C’est une question d’éthique, selon lui.
Le 10 novembre, Pfizer a envoyé une lettre aux participants, indiquant que la société étudie des moyens de permettre aux participants intéressés du groupe placebo qui répondent aux critères d’éligibilité pour l’accès d’urgence de passer dans le groupe « vaccin » de l’essai. Un porte-parole a déclaré à la revue Nature que l’entreprise aurait « la responsabilité éthique d’informer tous les participants à l’étude de la disponibilité d’un vaccin d’urgence autorisé ».
Nature a entendu une douzaine de participants aux essais Pfizer-BioNTech ou Moderna, dont la plupart ont déclaré que s’ils apprenaient qu’ils avaient reçu un placebo, ils prendraient le vaccin s’il était offert. « Une des raisons pour lesquelles j’ai participé était que je comprenais que la norme pour les études en aveugle est de lever l’aspect aveugle si le vaccin est très efficace et d’offrir le vaccin à tous les groupes », explique Emma Bernay, participante à l’essai Moderna.
Mais si trop de personnes passent d’un groupe à l’autre, les essais pourraient ne pas avoir de groupes de contrôle suffisamment grands pour recueillir des résultats statistiquement significatifs pour certains objectifs à long terme, déclare Stöhr. Il s’agit notamment d’écarter tout problème de sécurité à long terme et d’établir de manière concluante si le vaccin empêche les personnes d’être infectées par le SRAS-CoV-2, ou s’il protège simplement les personnes infectées contre la maladie.


Il y a aussi le risque que des personnes participant à des essais autres que ceux de Pfizer, BioNTech et Moderna abandonnent leurs études pour se faire vacciner en vertu des dispositions d’utilisation d’urgence, explique Larry Corey, immunologiste au Fred Hutchinson Research Center de Seattle. Le porte-parole de Pfizer a déclaré que la société discutera avec la FDA de la manière dont elle rassemblera des données pour mesurer de manière exhaustive la sécurité et l’efficacité si les participants changent de groupe. Le plan d’essais cliniques de la société indique qu’elle a l’intention de surveiller les participants pendant deux ans après leur dernière dose de vaccin.
D’autres développeurs de vaccins sont également aux prises avec ces problèmes. Eduardo Spitzer, le directeur scientifique du laboratoire Elea Phoenix à Buenos Aires, qui mène les essais en Argentine d’un vaccin chinois de Sinopharm (Pékin), est convaincu que le pays lancera un programme de vaccination d’urgence. Si cela se produit, les médecins, les infirmières et autres travailleurs essentiels, dont beaucoup ont été inscrits à l’essai, pourraient recevoir des vaccinations obligatoires et ne seraient donc plus admissibles à participer à l’essai. D’autres participants du groupe placebo pourraient abandonner pour se faire vacciner.
Comment gérer les conséquences des autorisations d’urgence
Il existe des moyens de gérer de telles perturbations sans compromettre le résultat de l’essai, explique Kathleen Neuzil, directrice du Center for Vaccine Development and Global Health à l’Université du Maryland. Elle est également coprésidente du réseau d’essais de prévention COVID-19 des National Institutes of Health des États-Unis, qui organise des essais cliniques pour des entreprises telles que Pfizer et Moderna.
Les participants qui ont initialement reçu un placebo mais qui ont changé de groupe pour obtenir le vaccin pourraient être surveillés en tant que groupe séparé, et une comparaison de l’efficacité et de la sécurité à long terme du vaccin pourrait être faite entre ces groupes. Neuzil a utilisé une configuration similaire pour déterminer la durée de protection offerte par le premier vaccin contre le zona. Avant de lever l’aspect aveugle des essais, les entreprises pourraient également demander aux volontaires de rester dans l’étude et de se faire vacciner dès la fin de l’essai, explique Corey.
Christian Smerz de Houston, un participant à l’essai Pfizer, a déclaré à Nature qu’il comprenait l’importance du groupe placebo pour d’autres tests et qu’il envisagerait de rester dans l’essai. Les entreprises et les régulateurs peuvent également recueillir des données sur l’innocuité et l’efficacité des personnes appartenant aux groupes à haut risque qui achètent les vaccins, explique Eng Eong Ooi, chercheur en maladies infectieuses à la Duke – NUS Medical School à Singapour. Mais ces données peuvent être biaisées, car elles ne peuvent pas être comparées aux données d’un groupe témoin. Cependant, elles peuvent toujours fournir des informations utiles sur la sécurité et l’efficacité.
Néanmoins, une fois qu’un vaccin contre la COVID-19 reçoit une autorisation d’urgence, les essais de vaccins ultérieurs deviendront plus compliqués, explique Ooi, qui développe un vaccin qui en est aux premiers essais. Les entreprises qui lancent de nouveaux essais devront montrer que leurs vaccins sont meilleurs que ceux qui ont obtenu une approbation d’urgence, ce qui rend les essais plus coûteux. « Tout vaccin approuvé, même si ce n’est que pour une utilisation d’urgence, changera le paysage de la façon dont les vaccins arrivent sur le marché ».