
Ces dernières années, l’usage d’organoïdes a permis de réaliser de nombreuses avancées dans la recherche médicale. En effet, ces organes miniatures ont maintes fois permis aux scientifiques de mieux comprendre comment les maladies se développent et de tester diverses approches thérapeutiques. Une équipe de la faculté de médecine de l’Université de Californie à San Diego a récemment fait une nouvelle découverte majeure grâce à ces modèles d’organes : ils ont identifié la mutation génétique associée à une forme profonde d’autisme, puis ont réussi à restaurer tout le système neuronal en corrigeant cette mutation.
Plusieurs maladies neurologiques et neuropsychiatriques, y compris les troubles du spectre autistique (TSA) et la schizophrénie, ont été associées à des mutations du facteur de transcription 4 (TCF4), un gène du chromosome 18 essentiel au développement du cerveau et à la fonction neuronale ; les facteurs de transcription sont des protéines qui initient ou régulent la transcription des autres gènes. La façon dont les mutations pathologiques du TCF4 affectent les tissus neuronaux est cependant mal comprise.
Pour en savoir plus, des chercheurs ont examiné des neurones et des organoïdes cérébraux développés à partir de fibroblastes cutanés prélevés sur des enfants atteints du syndrome de Pitt-Hopkins, un trouble provoqué par certaines mutations spécifiques du TCF4. Le syndrome de Pitt-Hopkins est un TSA caractérisé par une déficience cognitive, une morphologie faciale distinctive, des problèmes gastro-intestinaux et des anomalies du rythme respiratoire. Grâce à l’observation des organoïdes, l’équipe a pu disséquer les mécanismes moléculaires pathologiques sous-jacents et caractériser les anomalies cellulaires résultant des mutations du TCF4.
Une mutation qui limite la prolifération et la différenciation des cellules
Les modèles murins du syndrome de Pitt-Hopkins ne parviennent pas à imiter avec précision les caractéristiques neuronales des patients atteints de ce trouble ; en convertissant les cellules cutanées prélevées sur de jeunes patients en cellules souches, les chercheurs ont pu fabriquer des cellules progénitrices neurales, des neurones et des « mini-cerveaux » reproduisant plus fidèlement les fonctions attendues du véritable organe. Ils ont ainsi pu suivre le développement des tissus comme s’ils examinaient la croissance d’un fœtus.
En comparant la croissance des tissus intégrant des versions mutées de TCF4 avec des tissus dotés de gènes TCF4 typiques, ils ont pu cartographier précisément les changements que les mutations occasionnaient dans la structure et le fonctionnement des tissus. « Même sans microscope, nous pouvions dire quel organoïde cérébral portait la mutation », a déclaré Alysson R. Muotri, directrice du programme de cellules souches de l’UC San Diego et auteure principale de l’étude.

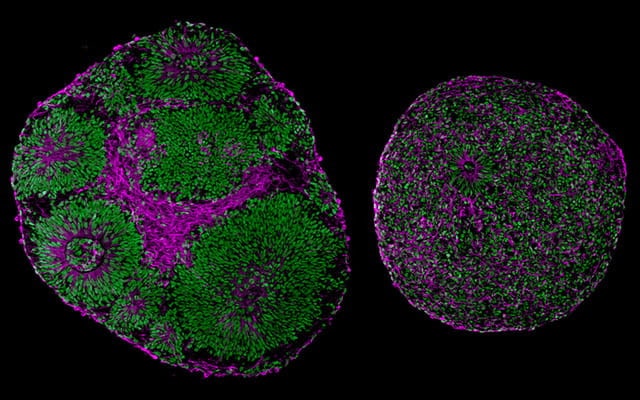 Des différences significatives de taille et de structure ont été observées entre les organoïdes cérébraux dérivés de patients atteints du syndrome de Pitt-Hopkins (à droite) et d’un organoïde normal (à gauche). © F. Papes et al.
Des différences significatives de taille et de structure ont été observées entre les organoïdes cérébraux dérivés de patients atteints du syndrome de Pitt-Hopkins (à droite) et d’un organoïde normal (à gauche). © F. Papes et al.Pour commencer, l’équipe a constaté que les organoïdes porteurs du syndrome étaient de taille et de structure aberrantes ; ils étaient sensiblement plus petits que les organoïdes normaux et contenaient un pourcentage plus élevé de cellules progénitrices neurales et beaucoup moins de neurones. En effet, les cellules progénitrices cultivées à partir des cellules souches présentaient une prolifération réduite et une moindre capacité à se différencier en neurones — ce qui suggère que la mutation du TCF4 inhibe la multiplication et la différenciation des cellules progénitrices neurales. Ces dernières affichaient par ailleurs une sénescence précoce.
Les quelques cellules qui étaient tout de même différenciées en neurones étaient moins actives que la normale et restaient regroupées au lieu de s’organiser en réseaux neuronaux. Selon les chercheurs, cette architecture atypique est probablement à l’origine de l’altération des fonctions cognitives et motrices dans le cas du syndrome de Pitt-Hopkins. Dès lors, ils se sont demandé s’il était possible d’inverser ces modifications structurelles en agissant directement sur l’expression du TCF4.
Une possible restauration des fonctions motrices et cognitives
Une série d’expériences a révélé que la mutation TCF4 entraînait une réduction de la signalisation Wnt/β-caténine et de l’expression des facteurs de transcription SOX — deux signaux moléculaires importants, qui régulent la multiplication des cellules embryonnaires, leur maturation en neurones et leur migration vers la région cérébrale prévue. La baisse de différenciation neuronale semblait donc liée à cette réduction de signalisation.

Les chercheurs ont alors entrepris de soutenir pharmacologiquement la signalisation Wnt (via un composé chimique nommé CHIR99021) : la procédure a permis de restituer une partie de la diversité et de l’activité neuronale aux organoïdes malades. En outre, la correction directe des mutations du TCF4 (par édition génétique) a inversé leurs effets : les organoïdes porteurs du syndrome sont devenus plus similaires aux organoïdes normaux utilisés comme témoins. « Le fait que nous puissions corriger ce gène et que tout le système neuronal se rétablisse, même au niveau fonctionnel, est incroyable », a déclaré Muotri.

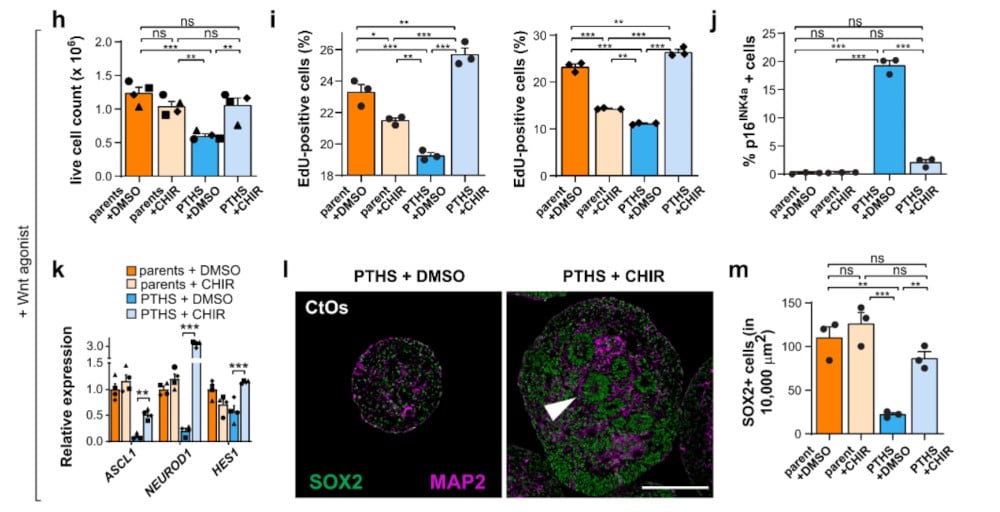 h) Comptage des cellules vivantes après traitement des cellules progénitrices neurales avec l’agoniste de la voie Wnt CHIR99021. j) Quantification des cellules sénescentes parmi les cellules progénitrices neurales traitées par CHIR99021. k) CHIR99021 restaure l’expression des gènes de prolifération dans les cellules traitées. l) Organoïdes porteurs du syndrome de Pitt-Hopkins à 4 semaines in vitro après traitement avec CHIR99021, montrant une augmentation marquée de l’abondance des cellules progénitrices neurales (en vert) et des neurones (en rose), ainsi que la réapparition des rosettes neurales (flèche). © F. Papes et al.
h) Comptage des cellules vivantes après traitement des cellules progénitrices neurales avec l’agoniste de la voie Wnt CHIR99021. j) Quantification des cellules sénescentes parmi les cellules progénitrices neurales traitées par CHIR99021. k) CHIR99021 restaure l’expression des gènes de prolifération dans les cellules traitées. l) Organoïdes porteurs du syndrome de Pitt-Hopkins à 4 semaines in vitro après traitement avec CHIR99021, montrant une augmentation marquée de l’abondance des cellules progénitrices neurales (en vert) et des neurones (en rose), ainsi que la réapparition des rosettes neurales (flèche). © F. Papes et al.L’équipe souligne néanmoins que ces manipulations ont eu lieu à un stade très précoce (prénatal) du développement du cerveau ; or, les enfants porteurs du syndrome de Pitt-Hopkins, comme d’autres TSA, ne reçoivent un diagnostic que vers l’âge de 2 ou 3 ans. Des essais cliniques doivent donc être menés pour vérifier si la même intervention peut être effectuée en toute sécurité et avec autant d’efficacité à cet âge. Muotri et ses collègues optimisent leurs outils de thérapie génique en vue de tels essais. « Pour ces enfants et leurs proches, toute amélioration de la fonction motrice-cognitive et de la qualité de vie vaudrait la peine d’être tentée », conclut la spécialiste.